Staphylococcus aureus FtsZ and PBP4 bind to the conformationally dynamic N-terminal domain of GpsB
- Department of Molecular Medicine, Morsani College of Medicine, University of South Florida, United States
- Department of Molecular Biosciences, University of South Florida, United States
- Department of Chemistry, University of South Florida, United States
- Global and Planetary Health, College of Public Health, University of South Florida, United States
Abstract
In the Firmicutes phylum, GpsB is a membrane associated protein that coordinates peptidoglycan synthesis with cell growth and division. Although GpsB has been studied in several bacteria, the structure, function, and interactome of Staphylococcus aureus GpsB is largely uncharacterized. To address this knowledge gap, we solved the crystal structure of the N-terminal domain of S. aureus GpsB, which adopts an atypical, asymmetric dimer, and demonstrates major conformational flexibility that can be mapped to a hinge region formed by a three-residue insertion exclusive to Staphylococci. When this three-residue insertion is excised, its thermal stability increases, and the mutant no longer produces a previously reported lethal phenotype when overexpressed in Bacillus subtilis. In S. aureus, we show that these hinge mutants are less functional and speculate that the conformational flexibility imparted by the hinge region may serve as a dynamic switch to fine-tune the function of the GpsB complex and/or to promote interaction with its various partners. Furthermore, we provide the first biochemical, biophysical, and crystallographic evidence that the N-terminal domain of GpsB binds not only PBP4, but also FtsZ, through a conserved recognition motif located on their C-termini, thus coupling peptidoglycan synthesis to cell division. Taken together, the unique structure of S. aureus GpsB and its direct interaction with FtsZ/PBP4 provide deeper insight into the central role of GpsB in S. aureus cell division.
Editor's evaluation
This valuable work reports a unique N-terminal motif of Staphylococcus aureus GpsB, the co-crystal structure of GpsB with the C-terminus of PBP4, and the direct interaction between GpsB and the C-terminus of FtsZ. The evidence supporting these discoveries is convincing, with biochemical and structural characterizations. This study sheds light on the role of GpsB in the cell division of this important pathogen.
https://doi.org/10.7554/eLife.85579.sa0Introduction
Bacterial cell division is a dynamic process involving more than a dozen proteins that form a multimeric complex at mid-cell. Collectively known as the divisome, this network of scaffolding proteins and enzymes stimulates peptidoglycan synthesis, constricts the existing membrane, and forms the septal cell wall (Mahone and Goley, 2020). While divisomal proteins may differ among certain clades, most are conserved among all bacteria. Perhaps the most important and well-studied divisomal protein is FtsZ, a bacterial homolog of eukaryotic tubulin. FtsZ marks the division site by forming a ‘Z-ring’ in association with early-stage divisomal proteins such as FtsA, ZapA, SepF, and EzrA in Gram-positive bacteria (Gamba et al., 2009; Lutkenhaus et al., 2012). Late-stage divisomal proteins such as DivIVA, FtsL, DivIB, and various penicillin-binding proteins (PBPs) subsequently assemble to carry out cell division and facilitate the separation and creation of identical daughter cells.
GpsB is a DivIVA-like protein that is highly conserved in Firmicutes (Halbedel and Lewis, 2019; Hammond et al., 2019). While GpsB is dispensable, or conditionally essential in most Firmicutes (Rismondo et al., 2016; Fleurie et al., 2014; Claessen et al., 2008; Tavares et al., 2008), it was reported to be essential for growth in Staphylococcus aureus (Santiago et al., 2015; Gillaspy, 2006). However, recent studies indicate that gpsB may not be essential, but nevertheless plays a significant role in maintaining S. aureus cell morphology (Sutton et al., 2023; Bartlett et al., 2023; Costa et al., 2023). In particular, the importance of S. aureus GpsB (Sa GpsB) for cell division is underscored by its unique ability to regulate FtsZ polymerization in S. aureus (Eswara et al., 2018). At this time, GpsB-FtsZ interaction has not been reported in other bacteria. Sa GpsB has also been reported to interact with other cell division proteins such as EzrA (Steele et al., 2011), DivIVA (Bottomley et al., 2017), and the wall teichoic acids (WTA) biosynthesis/export proteins TarO and TarG (Kent, 2024; Hammond et al., 2022).
The interaction of GpsB with PBPs has also been investigated in several bacteria, including Listeria monocytogenes (Lm) PBPA1, Streptococcus pneumoniae (Sp) PBP2a, and B. subtilis (Bs) PBP1 (Halbedel and Lewis, 2019). These studies show that GpsB binds to a short, ~5- to 30-residue N-terminal sequence found on the cytosolic region of these bitopic PBPs, referred to as an N-terminal ‘mini-domain’ (Rismondo et al., 2016; Cleverley et al., 2019). Cleverley et al. found PBP mini-domains containing an (S/T)-R-X-X-R-(R/K) motif directly interact with the N-terminal domain of GpsB by forming electrostatic interactions and hydrogen bonds with a shallow, acidic cavity located at the GpsB dimer interface (Figure 1—figure supplement 1A–C) Cleverley et al., 2019.
While Lm, Sp, and Bs have at least six annotated PBPs (Kocaoglu et al., 2015; Korsak et al., 2010; Kunst et al., 1997), there are only four in S. aureus: PBP1, PBP2, PBP3, and PBP4 (Gillaspy, 2006) - five in methicillin-resistant S. aureus (MRSA) which expresses an additional β-lactam insensitive PBP, PBP2a (Hartman and Tomasz, 1984). S. aureus is further distinguished by its highly cross-linked peptidoglycan and can readily become resistant to β-lactams via PBP4 and the acquisition of PBP2a (Łeski and Tomasz, 2005; Snowden and Perkins, 1990). It is believed this β-lactam-resistant phenotype relies on WTA assembly, which influences the function and localization of PBP4 and PBP2a (Atilano et al., 2010). While PBP2a is a historically recognized element of antibacterial resistance, PBP4 has recently been found to contribute to β-lactam insensitivity (Chatterjee et al., 2017). As the sole class C PBP in S. aureus, PBP4 bears the fold and architecture of a carboxypeptidase, but uniquely catalyzes both transpeptidase and carboxypeptidase reactions (Finan et al., 2001; Henze and Berger-Bächi, 1995; Navratna et al., 2010; Wyke et al., 1981).
In this report, we show that Sa GpsB directly binds to FtsZ and PBP4 through a signature GpsB recognition sequence. Further analysis of the GpsB N-terminal domain reveals unique conformations and innate flexibility that is integral to the function of GpsB. Together, these findings provide insight into the unique role of GpsB in synchronizing FtsZ dynamics with cell wall synthesis during cell division in S. aureus.
Results
The crystal structure of Sa GpsB N-terminal domain reveals an atypical asymmetric dimer
The full-length Sa GpsB is a relatively small protein of 114 residues. Its N-terminal domain homodimerizes as a coiled-coil, while its smaller C-terminal domain homotrimerizes, forming a hexamer as the biological unit (Halbedel and Lewis, 2019; Rismondo et al., 2016; Cleverley et al., 2016). Using X-ray crystallography, we solved the structure of the N-terminal domain (residues 1–70) of Sa GpsB at 1.95 Å resolution in the P21 space group (Figure 1A; Supplementary file 1), with four monomers per asymmetric unit forming two dimers (dimers A and B). The overall structure of GpsB is similar to previously determined GpsB orthologs (Figure 1—figure supplement 1; Rismondo et al., 2016; Cleverley et al., 2019) and retains the same fold of DivIVA (Oliva et al., 2010), though it shares much less sequence similarity (Figure 1E). Dimerization of the N-terminal domain is facilitated by a pattern of nonpolar residues every three to four residues, promoting the formation of a hydrophobic core in the coiled-coil. This N-terminal domain is partitioned into three regions: a relatively short α-helix with approximately two turns (residues 10–16), a second longer α-helix (residues 28–70) that forms a coiled-coil, and an amphipathic 10-residue loop (residues 17–27) that links these two helices and intertwines with the adjacent protomer, ‘capping’ GpsB (Figure 1B). This loop region is proposed to interact with the inner leaflet of the cell membrane (Halbedel and Lewis, 2019).
Figure 1 with 3 supplements see all
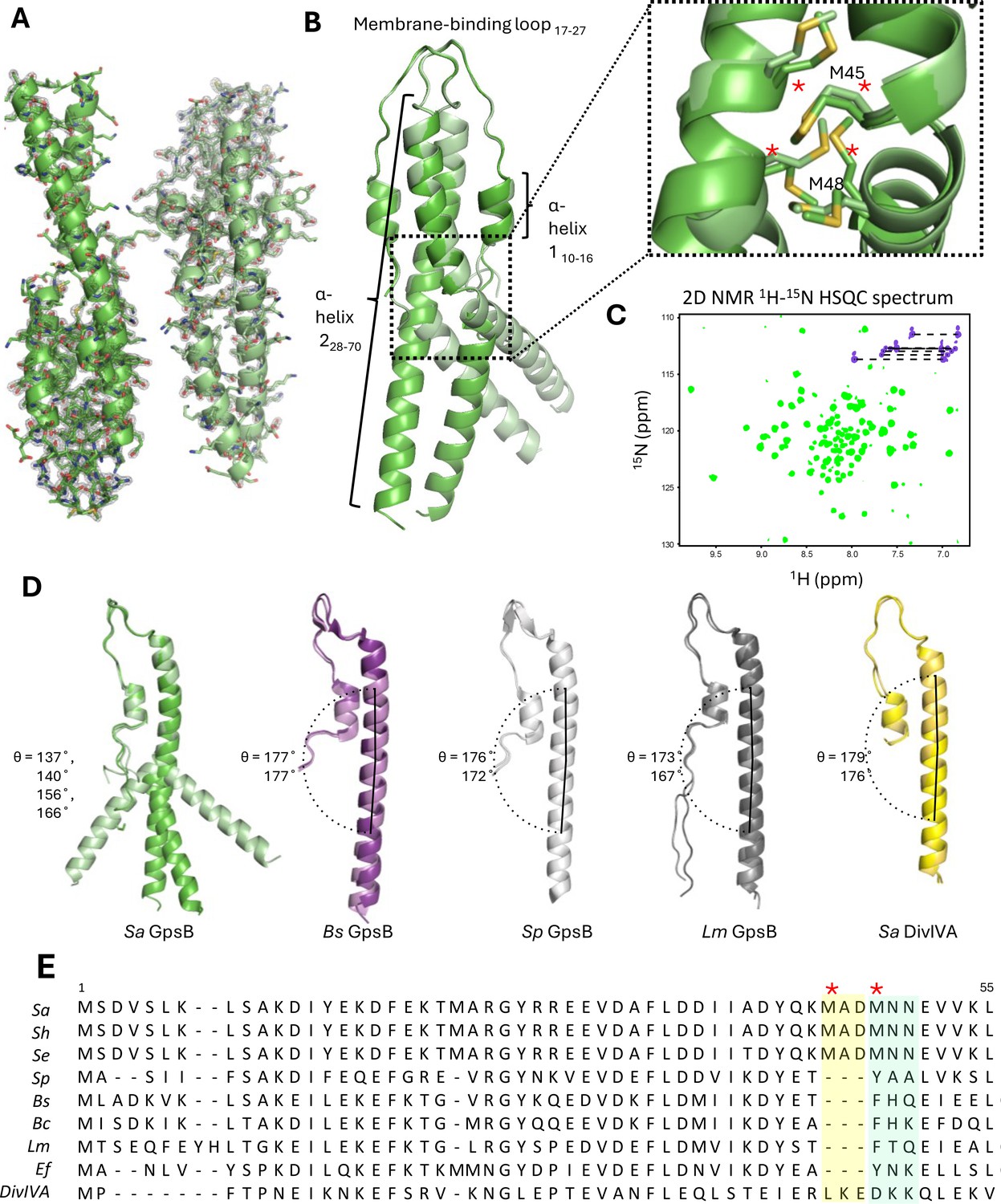
Crystal structure of the N-terminal domain of Sa GpsB.
(A) Two dimers lie antiparallel in the asymmetric unit from the P21 space group. The 2Fo-Fc electron density map, shown in gray, is contoured at 1σ with a resolution of 1.95 Å. (B) Superimposition of GpsB dimers reveals the dimers splay at a hinge region, formed by a cluster of four interlocked Met sidechains. *designates position in multisequence alignment shown in panel E. (C) The 1H-15N HSQC spectrum of the GpsB N-terminal domain, Sa GpsBWT1-70, shows a significantly higher number of signals compared to what is expected of a symmetric dimer based on the sequence of the domain, indicating the presence of conformational heterogeneity. Backbone and Asn/Gln sidechain amide signals are shown in green and purple respectively. (D) Comparison of different GpsB/DivIVA monomers from previously solved structures. Pitch angles were determined by placing a marker atom at the centroid of the beginning, midpoint ‘hinge’, and end of each helix, then measuring the angle. (E) Multisequence alignment of GpsB within select members of the Firmicutes phylum and with S. aureus DivIVA. Staphylococci GpsB contain a three-residue insertion that forms the hinge region, either MAD or MNN, depending on the sequence alignment parameters. The two Met residues (four per dimer) of the hinge region are designated with a red *. Sa - S. aureus, Sh - S. haemolyticus, Se - S. epidermidis, Sp - S. pneumoniae, Bs - B. subtilis, Bc - B. cereus, Lm - L. monocytogenes, Ef - E. faecalis, DivIVA - S. aureus DivIVA.
The most unique structural feature of Sa GpsB is a hinge forming at the midpoint of its N-terminal domain that causes each protomer to bend, adopting pitch angles (θ) of 137–140° (dimer A) and 156–166° (dimer B). In contrast, GpsB protomers from Lm, Bs, Sp, and Sa DivIVA are almost linear (θ=167–179°) and practically indistinguishable when superimposed (Figure 1D). We find that there is a 3-amino acid insertion in the Sa GpsB sequence where the helicity is disrupted and that a cluster of four Met residues (Met45, Met48) are interlocked at the dimer interface at this position (Figure 1B; dotted rectangle). These Met residues are only found in Staphylococci (Figure 1E) and take the place of an aromatic Tyr or Phe present in other orthologs, which normally stabilize the core via π stacking interactions. While the 3-amino acid insertion likely disrupts the continuity of the coiled-coil, methionine is one of the most flexible aliphatic amino acids and may further contribute to the conformational flexibility.
Experiments using solution state NMR spectroscopy further support the notion of intrinsic conformational heterogeneity, suggesting it is a bona fide structural feature rather than a crystallization artifact. The Sa GpsBWT1-70 construct contains a two-residue N-terminal extension (GH) after removal of the purification tag, and thus a total of 72 signals are expected. The 2D 1H-15N HSQC spectrum shows good signal dispersion (Figure 1C), which is characteristic of a folded domain. However, a total of 100 well-resolved backbone amide signals are observed, and seven pairs of signals are detected in the Asn/Gln sidechain region instead of four pairs (Figure 1C, Figure 1—figure supplement 2B). The appearance of the additional signals is not due to proline cis/trans isomerization since there are no prolines in the sequence, nor is it due to self-aggregation of the dimers to form a dimer-of-dimers or higher order oligomers, because raising the concentration from 160 to 700 μM has no effect on the number of signals in the spectrum (Figure 1—figure supplement 2B). In addition, the 1H-15N HSQC of Sa GpsBWT1-70 exhibits differential linewidths, with a set of 13 narrow signals at the center of the spectrum where disordered segments appear, and a larger set of 87 signals dispersed throughout the spectrum. The number of the narrow signals corresponds well with the number of residues found at the flexible N-terminus, suggesting that the appearance of extra signals is due to sampling of multiple conformations of the coiled-coil. Furthermore, three of the four Asn/Gln sidechain pairs showing chemical shift degeneracy are adjacent to the hinge (Figure 1—figure supplement 2A) providing strong evidence that the observed conformational heterogeneity occurs at this region.
Molecular dynamics (MD) simulations using Gromacs v. 5.0.4 and a CHARMM36m force field also support the idea that conformational flexibility exists in solution. After approximately 100 ns, dimer 1 adopts an ~180° pitch angle that occurs concomitantly to major fluctuations in dimer 2 in a separate simulation (Figure 1—figure supplement 3A), although the preference for continuous helical structures (corresponding to ~180° pitch angles) may sometimes be influenced by specific force field parameters.
A three-residue insertion unique to Staphylococci GpsB disrupts the coiled-coil pattern and destabilizes the structure of Sa GpsB
A unique element of Sa GpsB that likely contributes to its distinct conformational flexibility are three extra residues located at the hinge region - MAD or MNN (depending on the alignment parameters; Figure 1E), roughly corresponding to an extra turn in the helix. Deleting either MAD or MNN produces a homology model where residues 45–70, normally displaced by one turn, are now aligned with their orthologous residue pair (Figure 1—figure supplement 3B). To investigate the relative degree of stability imparted by these residues, GpsB mutant lacking the extra MAD or MNN residues at the hinge region was recombinantly constructed and purified, and Tm (melting temperature) was determined using circular dichroism (CD) spectroscopy (Figure 2A). This experiment shows the ΔMAD and ΔMNN GpsB have superior thermal stability to wildtype (WT) Sa GpsB for both the N-terminal domain (1–70) and full-length constructs. Perhaps the most notable finding from this experiment was that, while the Tm of Sa GpsBΔMAD1-70 (38.88 ± 0.18°C) was only modestly higher than Sa GpsBWT1-70 (34.63 ± 0.33°C), the Tm of Sa GpsBΔMNN1-70 (53.65 ± 0.55°C) was highly stable, approximately 1.5-fold higher than that of Sa GpsBWT1-70 and comparable to the thermal stability of the full-length Sa GpsBWTFL. Though the full-length Sa GpsBΔMNNFL was not analyzed, we expect that its Tm would be higher than both Sa GpsBWTFL and Sa GpsBΔMADFL based on the experiments assessing the 1–70 constructs.
Figure 2 with 2 supplements see all
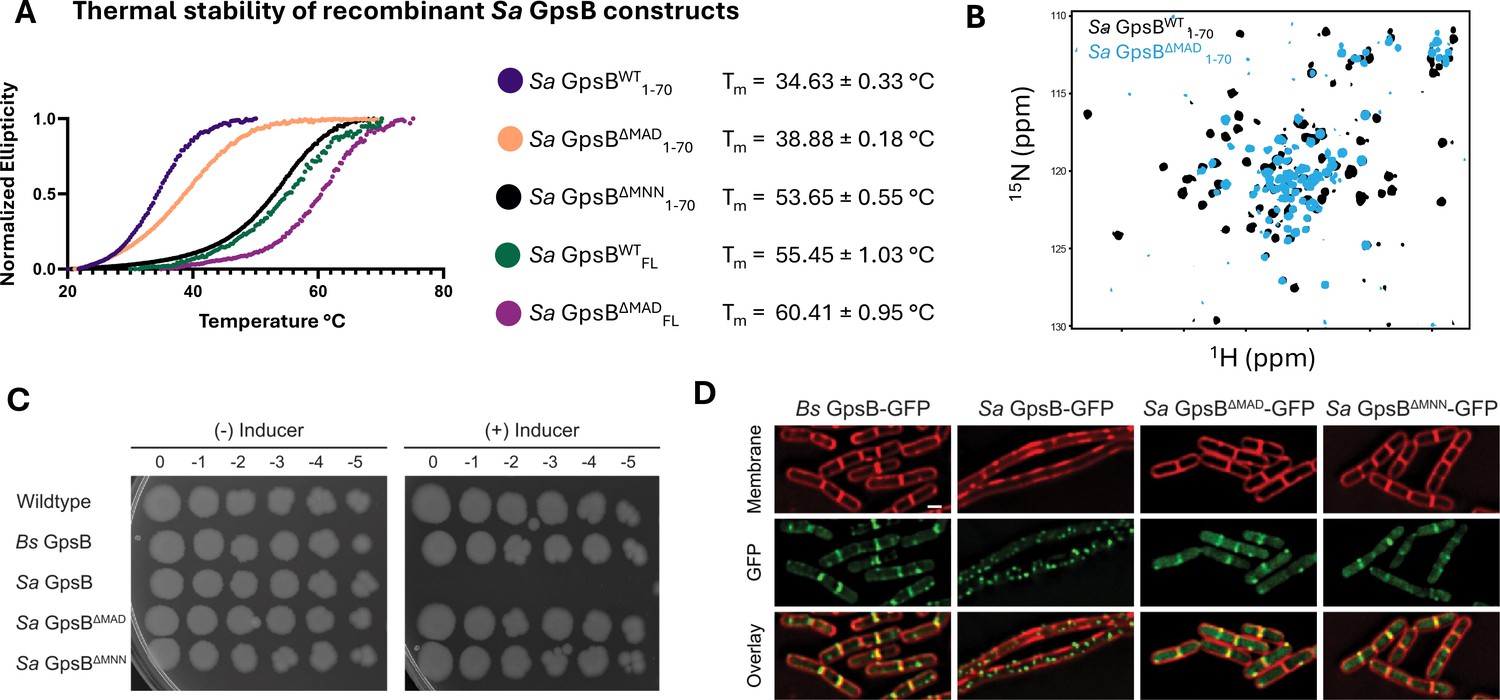
Deletion of a three-residue insertion in Sa GpsB increases thermal stability in solution and abolishes toxicity in B. subtilis.
(A) Circular dichroism (CD) melt profiles of recombinantly expressed Sa GpsB constructs reveal ΔMAD and ΔMNN mutants have increased Tm, compared to wildtype (WT) Sa GpsB. (B) Overlays of the 1H-15N HSQC of spectrum of Sa GpsBWT 1-70 and Sa GpsBΔMAD 1-70 suggest there are significant differences in the conformational properties of these two proteins. (C) Serial dilutions of B. subtilis strains harboring inducible Bs GpsB (GG18), Sa GpsB (GG7), Sa GpsB ∆MAD (LH119), and Sa GpsB ∆MNN (LH115), plated on LB plates without (left) and with (right) 1 mM IPTG demonstrate WT Sa GpsB is lethal, but ΔMAD Sa GpsB and ΔMNN Sa GpsB are not. (D) Fluorescence micrographs showing the protein localization of Bs GpsB-GFP (GG19), Sa GpsB-GFP (GG8), Sa GpsB-GFP ∆MAD (LH126), and Sa GpsB-GFP ∆MNN (LH116). Cell membrane was visualized using SynaptoRed membrane dye (1 µg/mL). Scale bar, 1 µm. In contrast to WT Sa GpsB, strains of B. subtilis that overexpress ΔMAD and ΔMNN Sa GpsB have similar cellular morphology to WT B. subtilis and these proteins localize to the division septum.
The conformational properties of the Sa GpsBΔMAD1-70 mutant were also different when analyzed by 1H-15N HSQC (Figure 2B). Most of the dispersed signals from the coiled-coil region experience severe broadening, in many cases beyond detection, as well as changes in chemical shift, while all narrow signals at the center of the spectrum show no change in linewidth or positions. Signal broadening is caused by changes in the rate of interconversion between the available conformations from the intermediate-slow for WT (s) to the intermediate-fast exchange regime for ΔMAD (ms), but without altering the overall number of states, as seven pairs of Asn/Gln sidechain pairs of signals are observed. Conformational rigidity is a well-established correlate of thermal stability (Karshikoff et al., 2015), and may contribute to the enhanced Tm of Sa GpsBΔMNN and Sa GpsBΔMAD. The aforementioned homology model (Figure 1—figure supplement 3B) corresponding to Sa GpsBΔMNN and Sa GpsBΔMAD reveals several residues potentially form stronger interactions than the WT. These include an intrahelical electrostatic interaction that replaces a potential repulsion between Asp47 and Glu51 with Asn47/Lys51 in Sa GpsBΔMAD and Asp47/Lys51 in Sa GpsBΔMNN (Figure 1—figure supplement 3C). The favorable Asp47/Lys51 electrostatic interactions in GpsBΔMNN may further contribute to its higher thermostability.
The MAD/MNN insertion is critical for GpsB function
Previously, we reported that overproduction of Sa GpsB in B. subtilis causes cell division arrest which eventually leads to filamentation and cell lysis (Eswara et al., 2018). We used this system to probe the significance of the flexibility provided by MAD/MNN residues for the function of GpsB. First, we conducted a growth assay on solid medium by spotting serial dilutions of cells of B. subtilis WT and cells harboring an inducible copy of Bs gpsB, Sa gpsB, Sa gpsBΔMAD, or Sa gpsBΔMNN. As previously established, overproduction of Bs GpsB is not lethal, but Sa GpsB is (Figure 2C; Eswara et al., 2018; Hammond et al., 2022). In contrast, overproduction of either ΔMAD or ΔMNN Sa GpsB is not lethal and cells grow as well as the negative controls (B. subtilis WT and inducible Bs GpsB strain). These results suggest the hinge region of Sa GpsB, characterized by two Met residues (Figure 1B), is essential for the normal function of Sa GpsB as assessed by lethal phenotype in B. subtilis. To further investigate the impact of these mutations, we examined GFP tagged ΔMAD and ΔMNN Sa GpsB in B. subtilis using high-resolution fluorescence microscopy. As reported previously, and as shown in Figure 2D, Bs GpsB-GFP localizes to the division site (Claessen et al., 2008; Tavares et al., 2008; Hammond et al., 2022) and does not lead to filamentation upon overproduction (2.09 ±0.49 µm; n=50), but Sa GpsB-GFP forms foci throughout the entire cell, and causes severe filamentation (22.83 ±16.51 µm; n=21) (Eswara et al., 2018). However, ΔMAD and ΔMNN variants of Sa GpsB-GFP do not cause filamentation (2.13 ±0.44 µm and 2.10 ±0.43 µm respectively; n=50) and are clearly localized at the division site. It has been noted previously that Sa GpsB-GFP also localizes to the division sites at initial stages prior to causing filamentation at a lower inducer concentration (Eswara et al., 2018). Taken together, this data suggests the ΔMAD and ΔMNN mutants presumably interact with the B. subtilis cell division machinery to allow for division site localization, but fail to elicit lethal filamentation and toxicity (Eswara et al., 2018).
We also investigated the phenotypes of S. aureus strains that overexpress Sa GpsBΔMAD and Sa GpsBΔMNN. First, we conducted a growth assay by spotting serial dilutions of S. aureus strains harboring an empty vector (EV) or an additional plasmid-based inducible copy of Sa gpsB, Sa gpsBΔMAD, or Sa gpsBΔMNN. Overproduction of Sa GpsB leads to a 100- to 1000-fold growth inhibition compared to EV control (Figure 2—figure supplement 1A). Interestingly, while cells overproducing Sa GpsBΔMNN grew similarly to the EV control, Sa gpsBΔMAD overexpression resulted in growth inhibition comparable to Sa GpsB. We hypothesized that the difference in phenotype is due to differential affinity of native Sa GpsB (produced from chromosomal locus) to Sa GpsBΔMAD and Sa GpsBΔMNN mutants (produced from a plasmid-based system), leading to varying propensity for homo/hetero complex formation. To test this, we conducted bacterial two-hybrid analysis in MacConkey plates and liquid culture as reported previously (Hammond et al., 2022; Battesti and Bouveret, 2012). As shown in Figure 2—figure supplement 1B, the affinity of Sa GpsBΔMNN to itself appears to be greater compared to Sa GpsBΔMNN and Sa GpsB. Sa GpsBΔMAD appears to have similar affinity to itself and Sa GpsB, but it is weaker compared to the self-interaction of Sa GpsB. Thus, the differential affinity between mutant Sa GpsB and WT Sa GpsB could underlie the observed difference in phenotypes. Next, we analyzed the cell morphology of S. aureus strains overproducing ΔMAD/ΔMNN mutants. As we have previously shown, overproduction of Sa GpsB leads to cell size enlargement due to cell division inhibition (Eswara et al., 2018). Using fluorescence microscopy (Figure 2—figure supplement 1C and D), we re-confirmed the increase in cell diameter in cells overproducing Sa GpsB (1.00 ±0.19 µm) when compared to the EV control (0.90 ±0.13 µm). In agreement with the lethal plate phenotype (Figure 2—figure supplement 1A), cells overproducing Sa GpsBΔMAD also displayed a statistically significant increase in cell diameter (0.95 ±0.16 µm), while Sa GpsBΔMNN did not (0.91 ±0.15 µm) and resembled EV control. We also ensured the stable production of Sa GpsB, ΔMAD, and ΔMNN via western blotting (Figure 2—figure supplement 1E). Lastly, by GFP tagging, we observed that both mutants localize to the division site similar to Sa GpsB-GFP (Figure 2—figure supplement 1F; Eswara et al., 2018; Hammond et al., 2022). In summary, ΔMNN is less functional compared to ΔMAD in terms of causing growth inhibition on plate and cell enlargement phenotype, however both ΔMAD and ΔMNN mutants are able to localize to division sites.
To further test the functionality of the hinge region mutants, we generated a Sa gpsB knockout strain using CRISPR/Cas9-based approach (Chen et al., 2017). As the sole copy, Sa gpsBΔMAD appears to be less toxic compared to the inducible WT Sa gpsB, suggesting that it is partially functional (Figure 2—figure supplement 2A). However, the ability of Sa gpsBΔMNN to impact S. aureus growth is severely impaired and resembles the EV control. This observation was consistent with the cell diameter quantification of different strains (Figure 2—figure supplement 2B). We also ensured the stability of these mutants is unaffected via western blotting (Figure 2—figure supplement 2C). To probe whether these mutants inhibit the native function of Sa GpsB when in a heteromer, similar to other previously isolated and functionally defective Sa GpsB mutants (such as Sa gpsBΔLEE) (Hammond et al., 2022), we co-expressed Sa gpsB-gfp and Sa gpsB, Sa gpsBΔMAD, or Sa gpsBΔMNN in B. subtilis. We observed that co-expression of hinge region mutants does not completely abolish the toxicity of Sa gpsB-gfp (Figure 2—figure supplement 2D). This result suggests that, although the Sa gpsBΔMAD and Sa gpsBΔMNN mutants are by themselves nonfunctional in B. subtilis (Figure 2C), they do not drastically inhibit the function of WT GpsB in the heteromer. We hypothesize that the deletion mutants lacking the flexible hinge region may to some degree potentially mimic certain GpsB conformations with varied activities. Thus the new data may indicate that the protomers adopting different conformations within the GpsB complex can function independently of one another, providing a mechanism to fine-tune the function of the whole complex.
The C-terminus of Sa FtsZ binds to the N-terminal domain of Sa GpsB through a conserved (S/T/N)-R-X-X-R-(R/K) motif
One of the few proteins known to interact with Sa GpsB is the tubulin-like GTPase, FtsZ - a central cell division protein that marks the division site in nearly all bacteria. We previously demonstrated that GpsB directly interacts with Sa FtsZ to stimulate its GTPase activity and modulate its polymerization characteristics (Eswara et al., 2018). However, the molecular basis for this interaction was not known. Remarkably, we discovered that the last 12 residues of Sa FtsZ (N-R-E-E-R-R—S-R-R-T-R-R), also known as the C-terminal variable (CTV) region (Buske and Levin, 2012), are a repeated match of the consensus GpsB-binding motif (S/T-R-X-X-R-(R/K)) found in the N-termini of Bs PBP1, Lm PBPA1, and Sp PBP2a. In this instance, the first motif bears an Asn instead of a Ser/Thr. Given the similar physicochemical properties of Asn as a small polar amino acid, it can likely replace Ser or Thr without any functional significance. To our knowledge, the interaction between GpsB and FtsZ is unique to S. aureus (Hammond et al., 2019), which is consistent with the absence of this motif in FtsZ orthologs from other organisms (Figure 3A).
Figure 3
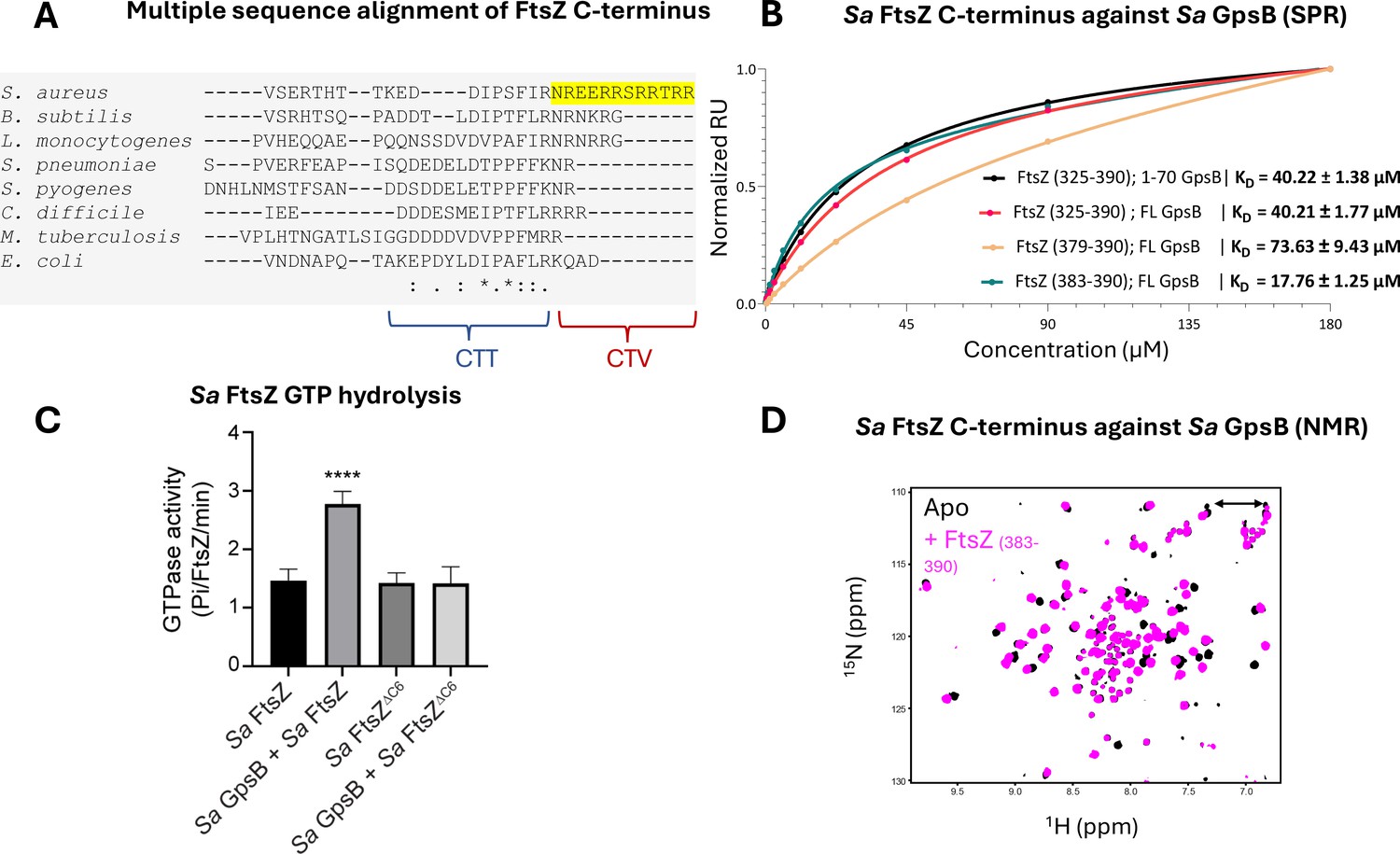
Sa FtsZ contains a repeated GpsB recognition motif at its C-terminus.
(A) A multiple sequence alignment of the FtsZ C-terminus from different representative bacteria reveals that there is a repeated GpsB recognition motif in Sa FtsZ (highlighted region) and that it is unique to this bacterium. (B) Surface plasmon resonance (SPR) titration of peptides corresponding to several segments of Sa FtsZ against Sa GpsB (residues 1–70 or full length). A titration of Sa FtsZ (residues 325–390) against 1–70 GpsB (black), corresponding to the N-terminal domain, shows binding can be isolated to this region. (C) When incubated with Sa GpsB, a Sa FtsZ mutant with a C-terminal truncation (SRRTRR, FtsZΔC6) has significantly lower GTP hydrolysis compared to its full-length counterpart. GTP hydrolysis was measured by monitoring inorganic phosphate (Pi) released (µmoles/min) by either FtsZ or FtsZΔC6 (30 μM) in the absence and presence of GpsB (10 μM). The plot is the average of n=6 independent data sets. p-Value for **** is <0.0001. (D) Overlays of the 1H-15N HSQC of spectrum of Sa GpsB (1–70) in the absence (black) and in the presence of FtsZ (383–390; pink). The boxed region highlights the only sidechain pair of signals that becomes affected by the addition of Ftsz. Based on a model derived from the structure of Bs GpsB in complex with a penicillin-binding protein (PBP)-derived peptide (Figure 1—figure supplement 2), it is tentatively assigned to Q43 of Sa GpsB.
To initially test whether the predicted FtsZ GpsB-binding motif directly binds to Sa GpsB, we purified and titrated the terminal 66 residues of Sa FtsZ (325–390) against full-length Sa GpsB using surface plasmon resonance (SPR), revealing a dose-dependent interaction (KD = 40.21 ± 1.77 µM; Figure 3B). A simultaneous titration against only the Sa GpsB N-terminal domain (1–70) demonstrates that the C-terminus of Sa FtsZ binds exclusively to this domain (KD = 40.22 ± 1.38 µM). This is further supported by assays with Sa FtsZ CTV (379–390; KD = 73.63 ± 9.43 µM) and the final eight residues of Sa FtsZ (383–390; KD 17.76±1.25 µM). The described biophysical affinity aligns with previous cellular studies (Cleverley et al., 2019), and is consistent with our hypothesis that the impetus for binding is the (S/T/N)-R-X-X-R-(R/K) motif which associates with the PBP-binding pocket (Figure 1—figure supplement 1). Unlike the previously identified GpsB-binding motifs located at the N-termini of PBPs, this is the first time such a motif has been found at the C-terminus of a GpsB-binding protein. The approximate fourfold difference in affinity between the CTV and the final eight residues could be a result of the inclusion of two Glu residues in the CTV (E381, E382), which may experience repulsion from the negatively charged PBP-binding pocket of GpsB. Notably, the GpsB recognition motif of Sa FtsZ is adjacent to the C-terminus carboxylate, which imparts an additional negative charge.
To further characterize the interaction between Sa FtsZ and Sa GpsB, we conducted a GTPase assay with Sa GpsB and Sa FtsZ or Sa FtsZ with the C-terminal six residues truncated (Sa FtsZΔC6). Briefly, in our previous report (Eswara et al., 2018), we found that Sa GpsB enhances the GTPase activity of Sa FtsZ. Therefore, we hypothesized that truncation of the terminal six residues of Sa FtsZ would eliminate the GpsB-mediated enhancement of GTPase activity. As shown in Figure 3C, and as reported previously, addition of Sa GpsB enhanced the GTPase activity of Sa FtsZ. However, this effect was not seen in Sa FtsZΔC6, suggesting that the last six C-terminal residues of Sa FtsZ is likely where the interaction with Sa GpsB occurs.
The interaction of the Sa GpsB N-terminal domain with the Sa FtsZ derived peptide (383–390) was also monitored using NMR (Figure 3D). Addition of the octapeptide to 15N-labeled N-terminal domain results in a significant chemical shift perturbation to a small number of dispersed signals in the 1H-15N HSQC spectrum, suggesting that the interaction is highly localized and occurs through the coiled-coil region without disturbing the conformational heterogeneity of the dimer. The available structures of Bs GpsB and Sp GpsB in complex with PBP-derived peptides (Figure 1—figure supplement 1A–C) suggest the complex is stabilized through interactions with helices 1 and 2, as well as part of the h1-h2 capping loop (Cleverley et al., 2019). The N-terminal Arg of Sa FtsZ octapeptide utilized in our experiments is expected to be placed in the PBP-binding pocket near Q43, based on the complex structures of GpsB homologs. Indeed, only one of the Asn/Gln pairs of signals in the 1H-15N HSQC is shifted upon addition of the peptide, while in agreement with our model all other sidechain signals lie far from the binding site (Figure 1—figure supplement 2A and B), suggesting that Sa GpsB recognizes partner proteins in a conserved manner.
The cytosolic, C-terminal mini-domain of Sa PBP4 binds to Sa GpsB through its (S/T/N)-R-X-X-R-(R/K) recognition motif
S. aureus has an unusually low number of PBPs in its genome (Gillaspy, 2006). Although PBP1, PBP2, PBP2a, and PBP3 all have a cytoplasmic N-terminal ‘mini-domain’, none bear a GpsB recognition motif (Figure 4A). Using SPR, we confirmed their cytoplasmic mini-domains have no affinity for Sa GpsB, which is in agreement with the findings from previous bacterial two-hybrid assays (Steele et al., 2011). Remarkably, PBP4, the only class C PBP encoded in the S. aureus genome, which purportedly functions as both a carboxypeptidase and transpeptidase PBP (Maya-Martinez et al., 2018), has a short, cytosolic C-terminal mini-domain with the sequence of N-R-L-F-R-K-R-K, satisfying the consensus GpsB-binding motif (S/T/N)-R-X-X-R-(R/K) found in S. aureus FtsZ and in orthologous PBPs found to bind to GpsB. Next, using SPR, we demonstrate this Sa PBP4 C-terminal octapeptide binds to Sa GpsB with a KD of 48.61±1.86 µM (Figure 4A). Supporting this finding is a potential interaction between Sa PBP4 and Sa GpsB previously noted in a bacterial two-hybrid study (Kent, 2024).
Figure 4 with 3 supplements see all
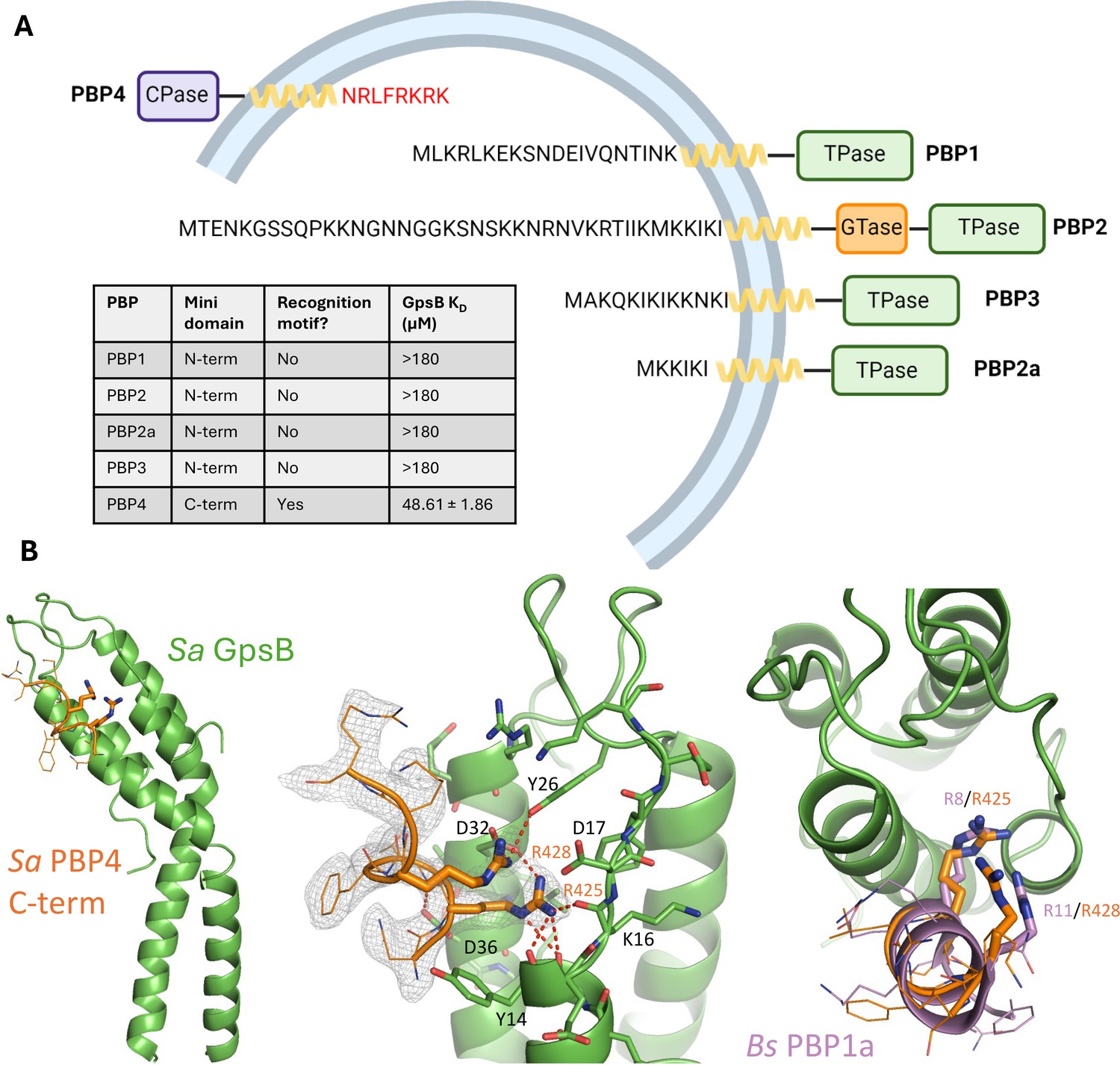
The C-terminal mini-domain of PBP4 directly interacts with GpsB.
(A) Domain representation of the four/five S. aureus (COL)/methicillin-resistant S. aureus (MRSA) (USA300) penicillin-binding proteins (PBPs). Each protein is shown from the N-terminus (left) to the C-terminus (right). All four transpeptidase PBPs - PBP1, PBP2, PBP3, and PBP2a - lack a GpsB recognition motif on their N-terminal, cytosolic mini-domain. In contrast, the C-terminal mini-domain of PBP4, the sole S. aureus class C PBP, contains this motif (NRLFRKRK, red). The dissociation constants were determined with SPR (n=2). (B) Crystal structure of Sa GpsB R24A in complex with PBP4 C-terminal peptide fragment at 2.40 Å resolution. The middle panel includes the electron density map of the Sa PBP4 heptapeptide, 2Fo-Fc=1.0σ. The right panel shows a superimposition of the Bs PBP1 mini-domain from the Bs GpsB+PBP1 complex (PDB ID 6GP7, purple) highlighting similar binding features.
Initial efforts to obtain a crystal structure of Sa PBP4 and Sa FtsZ with Sa GpsB were unsuccessful. A key factor preventing the formation of this complex were the tight interactions forming at the crystal packing interface. By extending the asymmetric unit, we found each GpsB dimer coordinates two others in a head-to-head arrangement (Figure 4—figure supplement 1A and B). This involves the insertion of Arg and Lys residues from the membrane-binding loop into the PBP-binding site from the adjacent GpsB protomer, mimicking the binding mode observed between PBP mini-domains and GpsB in orthologous structures, such as Bs PBP1 (Figure 4—figure supplement 1C and D; Cleverley et al., 2019). It is unclear whether this head-to-head interaction occurs in the cell or is simply a crystallization artifact. Nonetheless, it is apparent this interaction would need to be disrupted to capture interactions with a binding partner. To do so, we generated an R24A mutant because of its central role in the head-to-head interaction and its distance from the PBP-binding groove. This point mutation successfully disrupted the occluded crystal packing interface and allowed us to determine a 2.40 Å resolution structure of Sa PBP4 peptide bound to Sa GpsB (Figure 4B, Supplementary file 1). Unambiguous electron density for the Sa PBP4 C-terminal octapeptide, which adopted an α-helix, was resolved at the PBP-binding groove of GpsB. This structure clearly shows that two of these residues, Arg425 and Arg428, are key components of the Sa PBP4-Sa GpsB interaction, where they form multiple hydrogen bonds with the main chain amides of Sa GpsB Ile13, Tyr14, Lys16, and the sidechain hydroxyl of Tyr26. Furthermore, Arg425 and Arg428 form two salt bridges with the carboxyl sidechain of Asp32. Additionally, Sa GpsB Asp36 appears to play a major role in stabilizing the PBP4 α-helix by forming two hydrogen bonds with the backbone nitrogen of Arg425 and Leu426, an arrangement that is only possible when these two residues are part of an α-helix. Overall, the complex of Sa PBP4/Sa GpsB is very similar to Bs PBP1/Bs GpsB (Figure 1—figure supplement 1, Figure 4—figure supplement 1C and D) and is distinguished by the interactions of two Arg residues with the backbone and acidic sidechains lining the PBP-binding groove. We further used AlphaFold2-Multimer to study the interactions between Sa GpsB and FtsZ (Evans et al., 2021). Although attempts using full-length GpsB and/or long segments of the FtsZ C-terminal domain did not provide satisfying results, a model with the last six residues (S-R-R-T-R-R) was obtained and the nature of interaction resembles the Sa GpsB complex structure with the PBP4 peptide (Figure 4—figure supplement 3).
Sa FtsZ and Sa PBP4 have lower affinity for Sa GpsBΔMADFL compared to Sa GpsBWTFL
Due to the apparent importance of the three-residue insertion at the midpoint of Sa GpsB, we also tested the affinity of Sa FtsZ and Sa PBP4 derived peptides against Sa GpsBΔMADFL. Using SPR we found that the affinity was significantly reduced for Sa GpsBΔMADFL compared to Sa GpsBWTFL (Figure 4—figure supplement 2, Supplementary file 2). Dose-dependent saturation was very weak (KD>200 µM) for Sa PBP4 (423–431) and Sa FtsZ (379–390), which verged on being undetectable. Additionally, titration of 15N-labeled Sa GpsBΔMAD1-70 with the Sa FtsZ383-390 peptide does not result in chemical shift changes to the GpsBΔMAD 1H-15N HSQC, but only in broadening of a small number of signals, which is consistent with the higher KD measured by SPR (Figure 1—figure supplement 2C). While these mutants demonstrate better thermal stability (Figure 2A), it is possible their deletion may disrupt interactions with α-helix 1 (residues 10–16; Figure 1B), which may subsequently alter the size or shape of the PBP-binding pocket, thus reducing the favorable interactions that promote binding.
Discussion
GpsB is an important protein that coordinates multiple elements of the cell wall synthesis machinery. Although GpsB is widespread among Firmicutes, it has unique structural characteristics and functional roles in S. aureus. In this study, we initially present the crystal structure of the Sa GpsB N-terminal domain (1–70) (Figure 1A). The characteristic coiled-coil motif demonstrates conformational flexibility resulting from a three-residue hinge region that is unique to Staphylococci (Figure 1C, D, and E). The function of this hinge region and the flexibility it imparts remains unclear, but its deletion increases thermal stability (Figure 2A) and weakens affinity for Sa FtsZ and Sa PBP4 (Supplementary file 2). Furthermore, unlike WT Sa GpsB, ΔMAD and ΔMNN mutants are not toxic to B. subtilis (Figure 2C and D), underscoring the cellular significance of this region. In S. aureus, we show that the phenotypes of ΔMAD (but not ΔMNN) mimic those of Sa GpsB (Figure 2—figure supplement 1A and C). Regardless, both ΔMAD and ΔMNN mutants localize to the division site in both S. aureus and B. subtilis suggesting they retain some level of affinity for their usual interaction partners. In S. aureus ΔgpsB background, ΔMAD and ΔMNN mutants are less functional to varying extent (Figure 2—figure supplement 2A). Our analysis through co-expression of ΔMAD or ΔMNN with WT Sa GpsB in B. subtilis revealed that the heterocomplex is functional (Figure 2—figure supplement 2D). Taken together, we believe that the hinge region in Sa GpsB may permit the protomers with different conformations within the complex to function independently from each other to allow for the fine-tuning of function and/or protein interactions.
Next, we identified a GpsB recognition motif on the C-termini of both Sa FtsZ and Sa PBP4, leading to biophysical, biochemical, and structural experiments providing evidence of a direct interaction between these proteins and the N-terminal domain of Sa GpsB (Figure 3). This recognition motif, which is also present, but in the N-termini of Lm PBPA1, Sp PBP2a, and Bs PBP1, involves the insertion of several Arg residues into the binding groove formed at the coiled-coil interface near the membrane-binding loop (Figure 1—figure supplement 1; Cleverley et al., 2019).
Our group previously found that Sa GpsB interacts with Sa FtsZ to regulate its polymerization characteristics, but the repeated (S/T/N)-R-X-X-R-(R/K) GpsB recognition motif on its C-terminus was not recognized until the work on such motifs in PBPs published by Cleverley et al., 2019. This motif is unique to Staphylococci FtsZ and is absent/less conserved in other Firmicutes (Figure 3A). It is unclear why the GpsB recognition motif is repeated, since there is only enough area in the PBP-binding groove to accommodate a helix of eight to nine residues. It is possible that Sa FtsZ binds two Sa GpsB dimers simultaneously, given the putative arrangement of GpsB as trimer of dimers (Cleverley et al., 2016). Alternatively, the first site may be occluded by the binding of other FtsZ interaction partners that are known to bind FtsZ through the C-term such as FtsA, EzrA, and SepF (Huang et al., 2013). This finding, in addition to our previous reports (Eswara et al., 2018; Hammond et al., 2022), underscore the importance of GpsB in S. aureus cell division.
A crystal structure of the Sa PBP4 C-terminal recognition sequence bound to the N-terminal domain of GpsB reveals a binding mode that mimics previously solved orthologous PBP/GpsB pairs. This discovery is notable because it was previously thought that only the N-terminal ‘mini-domain’ of transpeptidase PBPs (class A, class B) could bind to GpsB. Furthermore, this finding is significant not only because it is the sole S. aureus PBP that binds to GpsB, but also because Sa PBP4 is intimately associated with WTA synthesis (Atilano et al., 2010; Farha et al., 2013). Previous studies have found that Sa GpsB interacts with the WTA biosynthesis pathway proteins TarO (Kent, 2024) and TarG (Hammond et al., 2022), and likely facilitates the export of WTA to the septal cell wall. To our knowledge, Sa PBP4 does not directly interact with any of the known WTA machinery. However, it does require WTA for recruitment to the division septum, and impairment of WTA assembly results in delocalization of Sa PBP4 (Atilano et al., 2010). Thus, while Sa PBP4 itself is not essential for growth, it is tightly regulated by WTA synthesis, an essential process that is also mediated by GpsB (Kent, 2024; Hammond et al., 2022). Thus, it is conceivable that GpsB likely arrives at the division site together with FtsZ, facilitates WTA synthesis, and subsequently recruits PBP4 to promote efficient cytokinesis.
PBP4 joins several other known proteins found to interact with Sa GpsB: EzrA (Steele et al., 2011), DivIVA (Bottomley et al., 2017), FtsZ (Eswara et al., 2018), TarO (Kent, 2024), and TarG (Hammond et al., 2022; Figure 5). The interaction diagram outlines the known interactions for GpsB to date but given the diverse number of proteins at the divisome and various binding surfaces on GpsB, it will surely be expanded in the future. Furthermore, while GpsB lacks certain direct interactions with other known divisome proteins, they are indirectly linked through intermediate proteins. For example, Sa FtsZ binds to Sa EzrA, a protein known to interact with the SEDS-PBP pairs Sa PBP1-FtsW and Sa PBP3-RodA, thus indirectly coupling Sa GpsB to enzymes that are critical for peptidoglycan synthesis at the divisome (Steele et al., 2011; Reichmann et al., 2019).
Figure 5
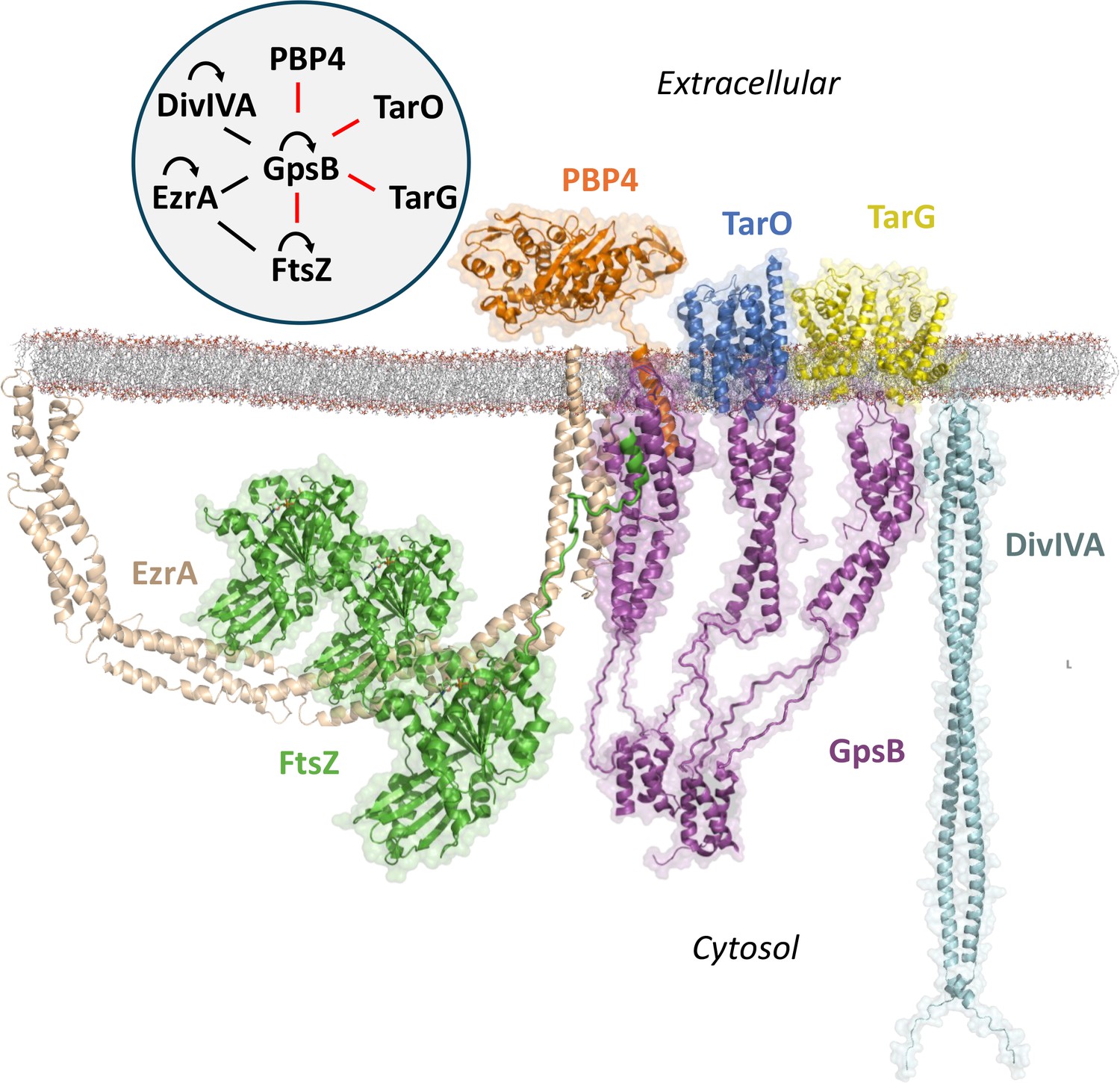
Known interactome of Sa GpsB and putative arrangement at the division septum in graphical and diagram (upper left) format.
In this paper, we demonstrate that the C-terminal mini-domain of PBP4 (orange) and the C-terminus of FtsZ (green) bind to the N-terminal domain of GpsB (purple). The regions within GpsB responsible for interacting with other partners remain to be elucidated. For interaction diagram, the red lines indicate interactions putatively unique to S. aureus, black lines indicate interactions found in both S. aureus and B. subtilis, and curved arrows represent self-interaction.
When evaluating the biophysical affinity of Sa FtsZ and Sa PBP4 for Sa GpsB, it is important to consider the peptide dissociation constants determined by SPR (~20–80 µM) may not directly translate to, and likely underestimate, the cellular affinity. The divisome is a highly complex environment with multiple proteins that interact near or at the cell membrane. The enrichment of both Sa FtsZ and Sa PBP4 at the division septum likely increases their apparent affinity simply based on avidity. Additionally, the arrangement of proteins may introduce synergistic interactions. Studies have found that EzrA, which binds to Sa GpsB (Steele et al., 2011), also binds to the C-terminus of FtsZ (Buske and Levin, 2012; Singh et al., 2007), likely upstream of the Sa GpsB recognition motif in S. aureus. This interaction could both increase the local concentration of Sa FtsZ and induce molecular recognition features that promote association with Sa GpsB. In addition, the oligomeric state of FtsZ and GpsB may further lead to cooperativity in the interactions between the FtsZ filament and GpsB hexamers. However, tighter binding may also prove deleterious in certain scenarios, especially for dynamic proteins like FtsZ. Given that Sa GpsB enhances the GTPase activity (required for FtsZ filament disassembly) of Sa FtsZ (Eswara et al., 2018), it is possible GpsB could dynamically promote polymerization and depolymerization of FtsZ.
Under the specter of growing antibacterial resistance, the identification of novel antibiotic targets is becoming increasingly urgent. Beyond PBPs, the bacterial divisome is a largely untapped source of antibiotic targets. The delineation of the physiological role of Sa GpsB, identification of its recognition motifs, and characterization of its 3D structure greatly enables modern antibiotic drug-discovery strategies. Furthermore, the involvement of Sa GpsB in multiple essential processes presents the opportunity to design targeted antibiotics. There is a growing need for narrow-spectrum antibiotics, especially for common infections, such as those caused by S. aureus (Melander et al., 2018). Narrow-spectrum antibiotics avoid selective pressure of commensal bacteria which can serve as a reservoir for resistance elements. In the same vein, their limited disruptive properties can avoid pathologies associated with bacterial dysbiosis, like Clostridioides difficile infection. The results from this study provide new information for both understanding the role of GpsB in S. aureus division and probing new avenues for narrow-spectrum antibiotic development.
Methods
Recombinant protein cloning and purification
The nucleotide sequence of Sa gpsB corresponding to the N-terminal domain (1–70) and full length (1–114) was inserted into a modified pET28a vector with a His-TEV sequence 5’ to the multiple cloning site (MCS). GpsB constructs expressed for SPR bioanalysis were cloned into a separate pET28a vector with a His-TEV-Avi sequence flanking the MCS (pAViBir). ∆MAD and ∆MNN mutants were generated with QuikChange site-directed mutagenesis using custom primers (∆MAD (5’-GATTATCAAAAAATGAATAATGAAGTTGTAAAATTATCAGAAGAGAATC) and ∆MNN (5’-GATTATCAAAAAATGGCCGATGAAGTTGTAAAATTATCAGAAGAG)). All plasmids were transformed into Rosetta (DE3) pLysS cells. A single colony was grown in LB media supplemented with 35 µg/mL chloramphenicol and 50 µg/mL kanamycin at 37°C overnight. The overnight culture was then diluted into 1 L LB media at 1:500 and incubated at 37°C until the OD600 reached 0.8. Protein expression was initiated with 0.5 mM IPTG and continued incubation at 25°C overnight. pAviBir constructs were biotinylated during IPTG induction with a stock of 5 mM biotin dissolved in bicine for a final concentration of 50 µM. Cells were harvested by centrifugation at 5000 × g for 10 min. The cell pellet was resuspended in buffer A (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 20 mM imidazole, and 10% glycerol). Cells were disrupted by sonication followed by centrifugation at 35,000 × g for 40 min. The pellet containing the protein was resuspended in buffer AD (100 mM Tris-HCl pH 8.0, 6 M guanidine HCl, 300 mM NaCl) and incubated at 30°C for ~1 hr to fully dissolve the pellet, followed by centrifugation at 45,000 × g for 1 hr. The supernatant was then loaded onto a HisTrap affinity column and eluted in a single step using Buffer BD (100 mM sodium acetate pH 4.5, 6 M guanidine HCl, 300 mM NaCl). The eluted protein was diluted dropwise in refolding buffer (100 mM Tris pH 8.0, 200 mM NaCl) and allowed to refold overnight at 4°C. The sample was then loaded to a HisTrap column and eluted with linear gradient of imidazole. The fractions containing GpsB were pooled and concentrated. The protein was then incubated with TEV at 1:20 ratio overnight at 4°C. The sample was then loaded onto a HisTrap for reverse Ni2+ cleanup. Flow-through was collected and purified using a HiLoad 16/60 Superdex 75 size exclusion column. The protein was stored at –80°C in the storage buffer (20 mM Tris-HCl pH 8.0, 200 mM NaCl). The purity of the protein was determined by SDS-PAGE as >95%. 15N-labeled GpsB (1-70) for NMR spectroscopy was expressed in the same way, but in minimal media containing 15N NH4Cl and 12C glucose as the source of nitrogen and carbon, respectively, supplemented with MgSO4, CaCl2, and trace metals.
Strain construction
Plasmids were generated using standard cloning procedures. The C-terminal six-residue truncation of FtsZ (FtsZ ΔC6) was generated by PCR using primer pairs oDB9/oDB10 (NdeI/XhoI) and cloned into the pET28a to create pDB1. FtsZ CTT encoding the C-terminal 66 amino acids of S. aureus FtsZ was cloned into pET28a using oP228/oP229 (Nde1/BamHI) to create pSK4. These were then transformed into BL21-DE3 cells creating EDB01 and SK7, respectively. ∆MAD was created by using site-directed mutagenesis with custom primers (5’-GATTATCAAAAAATGAATAATGAAGTTGTAAAATTATCAGAAGAGAATC) and ∆MNN was ordered from Integrated DNA Technologies as oLHgblock1. These mutations were then PCR-amplified and cloned into pDR111 to create both untagged (oP36/oP38; Hind111/Sph1) and GFP tagged (oP36/oP37; Hind111/Nhe1 and oP46/oP24; Nhe1/Sph1) variants. Plasmids were then transformed into PY79 and screened for amyE integration resulting in strains LH115, LH116, LH119, and LH126. The PCR products containing the ∆MAD and ∆MNN mutations were also cloned into pCL15 backbone using the same primers and restriction sites to create plasmids pLH62, pLH59, pLH63, and pLH60. These plasmids were then transformed into RN4220 cells resulting in strains LH129, LH127, LH130, and LH128. Then these plasmids were transduced into SH1000 to create strains LH135, LH134, LH133, and LH132. To make the BTH plasmids, ∆MAD and ∆MNN were amplified (BTH11/BTH12; EcoRI/XhoI) and cloned into pEB354 and pEB355 resulting in strains LH164, MA1, LH170, and LH168. The genotypes of strains and oligonucleotides used in this study are provided in Supplementary files 3 and 4.
CRISPR/Cas9-based gpsB deletion
Plasmid pCASSA was used to delete gpsB from S. aureus RN4220 genome (Chen et al., 2017). Guide RNA (gRNA) targeting gpsB was designed with primer pairs oLM25/26. Primers were phosphorylated via T4 polynucleotide kinase treatment. Following phosphorylation, primers were annealed in the same reaction mixture supplemented with 2.5 µL of 1 M NaCl at 95°C for 3 min. Annealed gRNA was inserted upstream of cas9 gene via compatible BsaI restriction sites utilized for Golden Gate Assembly. Upstream and downstream regions of gpsB were amplified for use in Gibson Assembly. The upstream region was amplified with primer pairs oLM27/28, containing restriction site XbaI compatible with pCASSA and complementary to downstream sequence. The downstream region was ordered in a geneblock (gLM1 with internal XhoI site corrected) and amplified with primer pairs oLM29/30, containing complementarity to upstream (Hammond et al., 2022) sequence and restriction site XhoI compatible with pCASSA. pCASSA containing gRNA was digested with XbaI and XhoI for linearization. Gibson Assembly was performed to insert amplified regions downstream of the gRNA scaffold. The resulting plasmid pLM29 was transformed into Escherichia coli DH5a. Correct conformation and insertion of sequences was confirmed via Sanger sequencing with primer pair oLM31/32. pLM29 was transformed into RN4220 via electroporation and incubated at 30°C for 24 hr. Colonies were screened with colony PCR for the amplification of gpsB. If gpsB was not detected, colony PCR was performed and the PCR product amplified using up- and downstream primers of gpsB locus was sent for Sanger sequencing to confirm gpsB deletion. Knockout strains were plasmid-cured by incubation of strains at 30°C overnight in 3 mL TSB. Overnight cultures were then diluted 1:1000 in TSB and incubated at 42°C until culture grew turbid. Confirmation of successful plasmid curing was determined by verifying susceptibility to 10 µg/mL chloramphenicol. Western blot analysis was also performed on knockout strains using α-GpsB antibody to ensure lack of GpsB production.
X-ray crystallography
Crystals of GpsB (1–70) were grown in a hanging drop apparatus by mixing 11 mg/mL GpsB (purity >98%) with crystallization buffer (30% PEG 3350, 0.4 M NaCl, and 0.1 M Tris pH 8.5) in an equal ratio at 20°C. Filamentous crystals appeared overnight and were harvested after 1 week of growth by briefly transferring to cryoprotectant (30% PEG 3350, 0.4 M NaCl, 0.1 M Tris pH 8.5, and 15% glycerol), followed by flash freezing in liquid nitrogen. GpsB (1–70) and PBP4 (424–451) were mixed in a 1:1 ratio with a GpsB concentration of 6 mg/mL and 1.44 mM PBP4 peptide (1:1 ratio). Initial crystals grew in 25% PEG 4000, 0.1 M Tris pH 8.0, and 0.2 M sodium acetate. These crystals were crushed, diluted 10,000-fold, then seeded into drops in a ratio of 1:1:0.5 (protein, crystallization solution, seed stock). Crystals were harvested after 1 week of growth by transferring to a cryoprotectant solution of 27.5% PEG 4000, 0.1 M Tris pH 8.0, 0.2 M sodium acetate, and 15% glycerol. X-ray diffraction data were collected on the Structural Biology Center (SBC) 19-ID beamline at the Advanced Photon Source (APS) in Argonne, IL, and processed and scaled with the CCP4 versions of iMosflm (Battye et al., 2011) and Aimless (Evans and Murshudov, 2013). Initial models were obtained using the MoRDa (Vagin and Lebedev, 2024) package of the online CCP4 suite. Unmodeled regions were manually built and refined with Coot (Emsley and Cowtan, 2004).
Circular dichroism
Thermal stability was assessed with CD using a Jasco J-815 CD spectropolarimeter coupled to a Peltier cell holder. Recombinantly expressed and purified WT GpsB and GpsB mutants were diluted to 2 μg/mL in 50 mM sodium phosphate (pH 7.0) and CD spectra were measured at 222 nm from 20°C to 80°C. Melting temperature was determined with a 4-parameter logistic curve fit using GraphPad Prism 9.
Surface plasmon resonance
A Series S CM5 chip (Cytiva) was docked into a Biacore S200 instrument (Cytiva) followed by surface activation with NHS/EDC amine coupling. Lyophilized neutravidin (Thermo Fisher Scientific) was dissolved in sodium acetate pH 5.25 to a final concentration of 0.25 mg/mL and injected onto the activated CM5 chip at 10 μL/min for 5 min. Biotinylated GpsB was diluted to 1 mg/mL in HEPES-buffered saline (HBS) and injected over the neutravidin-immobilized CM5 chip at 20 μL/min for 5 min. Synthetic peptides and recombinantly expressed Sa FtsZ (325–390) were serially diluted in HBS, and injected at 30 μL/min for 50 s with a dissociation of 100 s, followed by a stabilization period of 15 s and a buffer wash between injections. All experiments were performed in technical duplicate. Dissociation constants were determined with a one site binding model using GraphPad Prism 9.
NMR spectroscopy
2D 1H-15N HSQC spectra of Sa GpsB (1–70) were recorded on an Agilent 800-MHz direct drive instrument equipped with a cryoprobe. NMRpipe (Delaglio et al., 1995) and Sparky (University of California, San Francisco) were used for processing and analysis, respectively. All spectra were acquired in 20 mM Tris pH 8.0, 200 mM NaCl, prepared in 7.5% D2O, and at 25°C. The concentration of Sa GpsBWT1-70 was 160 or 700 μM for the free protein spectrum and 160 μM for the complex with the Sa FtsZ octapeptide, which was added in a 1.5× molar excess.
GTP hydrolysis of FtsZ
Sa FtsZ, Sa GpsB, and Sa FtsZΔC6 were purified using Ni-NTA affinity chromatography as described previously (Eswara et al., 2018). The effect of GpsB on the GTPase activity of FtsZ and FtsZΔC6 was determined by measuring the free phosphate released using the malachite green phosphate assay kit (Sigma-Aldrich, MAK307-1KT). Briefly, either FtsZ or FtsZΔC6 (30 μM) was incubated with GpsB (10 μM) in the polymerization buffer (20 mM HEPES pH 7.5, 140 mM KCl, 5 mM MgCl2) containing 2 mM GTP at 37°C for 15 min. The free phosphate released was determined by measuring the absorbance of the reaction mixture at 620 nm. Statistical analysis was completed using GraphPad Prism 9 with the one-way ANOVA and post hoc multiple comparisons.
B. subtilis and S. aureus growth conditions
Liquid cultures of B. subtilis cells were grown in LB and S. aureus cells were grown in TSB supplemented with 10 µg/mL chloramphenicol at 37°C.
Spot titer assays
Overnight cultures of B. subtilis and S. aureus strains were back diluted to OD600=0.1 and grown to midlog phase (OD600=0.4). Cultures were then back diluted to an OD600 of 0.1, serial diluted, and spotted onto LB plates with or without 1 mM IPTG (B. subtilis), or TSA plates supplemented with 10 µg/mL chloramphenicol with or without 1 mM IPTG (S. aureus), and incubated overnight at 37°C. Spot titers were repeated twice in technical triplicate each time.
Fluorescence microscopy
Overnight cultures of B. subtilis and S. aureus strains to be imaged were back diluted to OD600=0.1 and grown to midlog phase (OD600=0.4) and then induced with 1 mM IPTG and allowed to grow for an additional 2 hr. Cells were then prepared and imaged as previously described (Brzozowski et al., 2019). Briefly, 1 mL aliquots were spun down, washed, and resuspended in PBS. Cells were then stained with 1 μg/mL SynaptoRed fluorescent dye (MilliporeSigma, 574799-5MG) to visualize the membrane and 5 µL of culture was spotted onto a glass bottom dish (Mattek, P35G-1.5-14-C). Images were captured on a DeltaVision Core microscope system (Leica Microsystems) equipped with a Photometrics CoolSnap HQ2 camera and an environmental chamber. Seventeen planes were acquired every 200 nm and the data were deconvolved using SoftWorx software. Cells were measured using ImageJ and analyzed using GraphPad Prism 9 with the one-way ANOVA and post hoc multiple comparisons.
Bacterial two-hybrid assay
Plasmids carrying genes of interest cloned into the pEB354 (T18 subunit) and pEB355 (T25 subunit) backbones were transformed pairwise into BTH101 cells. Overnight cultures of the strains grown in 100 µg/mL ampicillin, 50 µg/mL kanamycin, and 0.5 mM IPTG, at 30°C, were spotted onto MacConkey agar containing 1% maltose that were also supplemented with ampicillin, kanamycin, and IPTG. Plates were incubated for 24 hr at 30°C and then imaged. Two biological replicates were performed each in technical triplicate. The β-galactosidase assay was carried out as previously described (Hammond et al., 2022). Mixtures of 20 µL of culture, 30 µL of LB, 150 µL Z buffer, 40 µL ONPG (4 mg/mL), 1.9 µL β-mercaptoethanol, and 95 µL polymyxin B (20 mg/mL) were transferred to a 96-well plate and read on a BioTek plate reader. Miller units were then calculated and graphed using GraphPad Prism 9 with the one-way ANOVA and post hoc multiple comparisons.
Immunoblot
Overnight cultures of S. aureus cells were back diluted to OD600=0.1, grown to midlog phase (OD600=0.4), and then induced with 1 mM IPTG and grown for an additional 2 hr. Cells were then standardized to an OD600=1.0, lysed with 5 µL lysostaphin (1 mg/mL in 20 mM sodium acetate), and incubated for 30 min at 37°C. 1 µL of DNAse A (1 U/µL) was added and incubated for an additional 30 min. Samples were then analyzed by SDS-PAGE analysis, transferred to a membrane, and probed with rabbit antiserum raised against GpsB-GFP. Total protein was visualized from the SDS-PAGE gel using the GelCode Blue Safe Protein Stain (Thermo Fisher, 24596).
Homology model construction of deletion variants
Template-based homology models were made using MODELLER 9.24 (Eswar et al., 2006) by constructing 1000 decoys corresponding to each construct based on various template structures.
MD simulations
The GpsB dimers were exposed to conventional MD simulation using Gromacs (v. 5.0.4) (Abraham et al., 2015; Páll et al., 2014) with the CHARMM36m force field (Huang et al., 2017). Explicit TIP3P water (Jorgensen et al., 1983) with 150 mM KCl was used for solvation. A 12 Å cut-off for the van der Waals forces was used. Electrostatic forces were computed using the particle mesh Ewald method (Darden et al., 1993). The Verlet cut-off scheme was used. The temperature and pressure were controlled using the Nosé-Hoover (Nosé, 1984b; Nosé, 1984a; Hoover, 1985; Parrinello and Rahman, 1981; Nosé and Klein, 1983) methods, respectively, to sample the NPT ensemble at P=1 bar and T=303.15 K. The integration time step was 2 fs, enabled by using H-bond restraints (Hess et al., 1977). Each system was simulated for 250–500 ns. All systems were made using CHARMM-GUI (Jo et al., 2008; Brooks et al., 2009; Lee et al., 2016).
AlphaFold2 predictions
Prediction of the complex between the GpsB dimer and the C-terminal domain of FtsZ was performed using AlphaFold2 (multimer version 3) (Evans et al., 2021; Varadi et al., 2022) on ColabFold (Mirdita et al., 2022; Steinegger and Söding, 2017). The sequence of GpsB was taken from S. aureus (strain COL, UniProt ID: Q5HFX8) and for FtsZ we used S. aureus (strain COL, UniProt ID: Q5HGP5) residues 325–390 for the entire C-terminal domain or residues 385–390 for the terminal 6-peptide. Structural predictions were made using all standard options on ColabFold and seeding the template structure resolved in this study between GpsB and the PBP4 fragment (PDB ID: 8E2C) was not observed to improve predictions.
Data availability
All crystal structures have been deposited in the RCSB Protein Data Bank (PDB) with accession IDs of: Sa GpsB NTD (PDB ID 8E2B), Sa GpsB NTD + Sa PBP4 C-term (PDB ID 8E2C).
-
RCSB Protein Data BankID 8E2B. N-terminal domain of S. aureus GpsB.
-
RCSB Protein Data BankID 8E2C. N-terminal domain of S. aureus GpsB in complex with PBP4 fragment.
References
-
iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLMActa Crystallographica. Section D, Biological Crystallography 67:271–281.https://doi.org/10.1107/S0907444910048675
-
Coordination of chromosome segregation and cell division in Staphylococcus aureusFrontiers in Microbiology 8:1575.https://doi.org/10.3389/fmicb.2017.01575
-
CHARMM: the biomolecular simulation programJournal of Computational Chemistry 30:1545–1614.https://doi.org/10.1002/jcc.21287
-
Live-cell fluorescence microscopy to investigate subcellular protein localization and cell morphology changes in bacteriaJournal of Visualized Experiments 01:59905.https://doi.org/10.3791/59905
-
Extreme C terminus of bacterial cytoskeletal protein FtsZ plays fundamental role in assembly independent of modulatory proteinsThe Journal of Biological Chemistry 287:10945–10957.https://doi.org/10.1074/jbc.M111.330324
-
PBP4 Mediates β-Lactam resistance by altered functionAntimicrobial Agents and Chemotherapy 61:e00932–e00917.https://doi.org/10.1128/AAC.00932-17
-
Rapid and efficient genome editing in Staphylococcus aureus by using an engineered CRISPR/Cas9 SystemJournal of the American Chemical Society 139:3790–3795.https://doi.org/10.1021/jacs.6b13317
-
Subunit arrangement in GpsB, a regulator of cell wall biosynthesisMicrobial Drug Resistance 22:446–460.https://doi.org/10.1089/mdr.2016.0050
-
Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systemsThe Journal of Chemical Physics 98:10089–10092.https://doi.org/10.1063/1.464397
-
NMRPipe: a multidimensional spectral processing system based on UNIX pipesJournal of Biomolecular NMR 6:277–293.https://doi.org/10.1007/BF00197809
-
Coot: model-building tools for molecular graphicsActa Crystallographica. Section D, Biological Crystallography 60:2126–2132.https://doi.org/10.1107/S0907444904019158
-
Comparative protein structure modeling using modellerCurrent Protocols in Bioinformatics 5:bi0506s15.https://doi.org/10.1002/0471250953.bi0506s15
-
How good are my data and what is the resolution?Acta Crystallographica. Section D, Biological Crystallography 69:1204–1214.https://doi.org/10.1107/S0907444913000061
-
Role of penicillin-binding protein 4 in expression of vancomycin resistance among clinical isolates of oxacillin-resistant Staphylococcus aureusAntimicrobial Agents and Chemotherapy 45:3070–3075.https://doi.org/10.1128/AAC.45.11.3070-3075.2001
-
Two-step assembly dynamics of the Bacillus subtilis divisomeJournal of Bacteriology 191:4186–4194.https://doi.org/10.1128/JB.01758-08
-
The Staphylococcus aureus NCTC 8325 genomeGram‐Positive Pathogens 01:381–412.https://doi.org/10.1128/9781555816513
-
Structural basis for interaction of DivIVA/GpsB proteins with their ligandsMolecular Microbiology 111:1404–1415.https://doi.org/10.1111/mmi.14244
-
Staphylococcus aureus penicillin-binding protein 4 and intrinsic beta-lactam resistanceAntimicrobial Agents and Chemotherapy 39:2415–2422.https://doi.org/10.1128/AAC.39.11.2415
-
A linear constraint solver for molecular simulationsJournal of Computational Chemistry 01:199709.https://doi.org/10.1002/(SICI)1096-987X(199709)
-
Canonical dynamics: Equilibrium phase-space distributionsPhysical Review A 31:1695–1697.https://doi.org/10.1103/PhysRevA.31.1695
-
FtsZ ring stability: of bundles, tubules, crosslinks, and curvesJournal of Bacteriology 195:1859–1868.https://doi.org/10.1128/JB.02157-12
-
CHARMM-GUI: a web-based graphical user interface for CHARMMJournal of Computational Chemistry 29:1859–1865.https://doi.org/10.1002/jcc.20945
-
Comparison of simple potential functions for simulating liquid waterThe Journal of Chemical Physics 79:926–935.https://doi.org/10.1063/1.445869
-
Rigidity versus flexibility: the dilemma of understanding protein thermal stabilityThe FEBS Journal 282:3899–3917.https://doi.org/10.1111/febs.13343
-
Profiling of β-lactam selectivity for penicillin-binding proteins in Streptococcus pneumoniae D39Antimicrobial Agents and Chemotherapy 59:3548–3555.https://doi.org/10.1128/AAC.05142-14
-
CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force fieldJournal of Chemical Theory and Computation 12:405–413.https://doi.org/10.1021/acs.jctc.5b00935
-
Bacterial cytokinesis: from z ring to divisomeCytoskeleton 69:778–790.https://doi.org/10.1002/cm.21054
-
Bacterial cell division at a glanceJournal of Cell Science 133:jcs237057.https://doi.org/10.1242/jcs.237057
-
ColabFold: making protein folding accessible to allNature Methods 19:679–682.https://doi.org/10.1038/s41592-022-01488-1
-
Constant pressure molecular dynamics for molecular systemsMolecular Physics 50:1055–1076.https://doi.org/10.1080/00268978300102851
-
A molecular dynamics method for simulations in the canonical ensembleMolecular Physics 52:255–268.https://doi.org/10.1080/00268978400101201
-
A unified formulation of the constant temperature molecular dynamics methodsThe Journal of Chemical Physics 81:511–519.https://doi.org/10.1063/1.447334
-
Features critical for membrane binding revealed by DivIVA crystal structureThe EMBO Journal 29:1988–2001.https://doi.org/10.1038/emboj.2010.99
-
ConferenceTackling exascale software challenges in molecular dynamics simulations with GROMACSInternational conference on exascale applications and software. pp. 3–27.https://doi.org/10.1007/978-3-319-15976-8
-
Polymorphic transitions in single crystals: a new molecular dynamics methodJournal of Applied Physics 52:7182–7190.https://doi.org/10.1063/1.328693
-
Peptidoglycan cross‐linking in Staphylococcus aureus: an apparent random polymerisation processJ European Journal of Biochemistry 191:373–377.https://doi.org/10.1111/j.1432-1033.1990.tb19132.x
-
Multiple essential roles for EzrA in cell division of Staphylococcus aureusMolecular Microbiology 80:542–555.https://doi.org/10.1111/j.1365-2958.2011.07591.x
-
MMseqs2 enables sensitive protein sequence searching for the analysis of massive data setsNature Biotechnology 35:1026–1028.https://doi.org/10.1038/nbt.3988
-
The roles of GpsB and DivIVA in Staphylococcus aureus growth and divisionFrontiers in Microbiology 14:1241249.https://doi.org/10.3389/fmicb.2023.1241249
-
Cytological characterization of YpsB, a novel component of the Bacillus subtilis divisomeJournal of Bacteriology 190:7096–7107.https://doi.org/10.1128/JB.00064-08
-
A role in vivo for penicillin-binding protein-4 of Staphylococcus aureusEuropean Journal of Biochemistry 119:389–393.https://doi.org/10.1111/j.1432-1033.1981.tb05620.x
Article and author information
Author details
Dipanwita Bhattacharya

Funding
National Institutes of Health (R21 AI164775)
- Yu Chen
- Prahathees Eswara
National Institutes of Health (R35 GM133617)
- Prahathees Eswara
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Eric Lewandowski for reading the manuscript. We also thank the staff members of the Advanced Photon Source of Argonne National Laboratory, particularly those at the SBC for assistance with X-ray diffraction data collection. SBC-CAT is operated by UChicago Argonne LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Copyright
© 2024, Sacco, Hammond et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,509
- views
-
- 213
- downloads
-
- 15
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 15
- citations for umbrella DOI https://doi.org/10.7554/eLife.85579
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Staphylococcus aureus FtsZ and PBP4 bind to the conformationally dynamic N-terminal domain of GpsB
eLife 13:e85579.
https://doi.org/10.7554/eLife.85579




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}