Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states
- The Victor Chang Cardiac Research Institute, Australia
- Nanyang Technological University, Singapore
- University of Oxford, United Kingdom
- The University of New South Wales, Australia
Abstract
A molecular model that provides a framework for interpreting the wealth of functional information obtained on the E. coli F-ATP synthase has been generated using cryo-electron microscopy. Three different states that relate to rotation of the enzyme were observed, with the central stalk’s ε subunit in an extended autoinhibitory conformation in all three states. The Fo motor comprises of seven transmembrane helices and a decameric c-ring and invaginations on either side of the membrane indicate the entry and exit channels for protons. The proton translocating subunit contains near parallel helices inclined by ~30° to the membrane, a feature now synonymous with rotary ATPases. For the first time in this rotary ATPase subtype, the peripheral stalk is resolved over its entire length of the complex, revealing the F1 attachment points and a coiled-coil that bifurcates toward the membrane with its helices separating to embrace subunit a from two sides.
https://doi.org/10.7554/eLife.21598.001eLife digest
ATP synthase is a biological motor that produces a molecule called adenosine tri-phosphate (ATP for short), which acts as the major store of chemical energy in cells. A single molecule of ATP contains three phosphate groups: the cell can remove one of these phosphates to make a molecule called adenosine di-phosphate (ADP) and release energy to drive a variety of biological processes.
ATP synthase sits in the membranes that separate cell compartments or form barriers around cells. When cells break down food they transport hydrogen ions across these membranes so that each side of the membrane has a different level (or “concentration”) of hydrogen ions. Movement of hydrogen ions from an area with a high concentration to a low concentration causes ATP synthase to rotate like a turbine. This rotation of the enzyme results in ATP synthase adding a phosphate group to ADP to make a new molecule of ATP. In certain conditions cells need to switch off the ATP synthase and this is done by changing the shape of the central shaft in a process called autoinhibition, which blocks the rotation.
The ATP synthase from a bacterium known as E. coli – which is commonly found in the human gut –has been used as a model to study how this biological motor works. However, since the precise details of the three-dimensional structure of ATP synthase have remained unclear it has been difficult to interpret the results of these studies.
Sobti et al. used a technique called Cryo-electron microscopy to investigate the structure of ATP synthase from E. coli. This made it possible to develop a three-dimensional model of the ATP synthase in its autoinhibited form. The structural data could also be split into three distinct shapes that relate to dwell points in the rotation of the motor where the rotation has been inhibited. These models further our understanding of ATP synthases and provide a template to understand the findings of previous studies.
Further work will be needed to understand this essential biological process at the atomic level in both its inhibited and uninhibited form. This will reveal the inner workings of a marvel of the natural world and may also lead to the discovery of new antibiotics against related bacteria that cause diseases in humans.
https://doi.org/10.7554/eLife.21598.002Introduction
In most cells, the bulk of ATP, the principal source of cellular energy, is synthesized by ATP synthase. This molecular generator couples ion flow across membranes with the addition of inorganic phosphate (Pi) to ADP thereby generating ATP (Iino and Noji, 2013; Stewart et al., 2014). Most bacteria, including Escherichia coli have only one type of rotary ATPase, referred to as F-type ATPase. Like the analogous complexes in other kingdoms, it is based on two reversible motors, termed F1 and Fo (Negrin et al., 1980), connected by central and peripheral stalks (Wilkens and Capaldi, 1998a) (Figure 1). The Fo motor spans the membrane converting the potential energy of the proton motive force (pmf) into rotation of the central stalk that in turn drives conformational changes in the F1 catalytic sites.
Figure 1
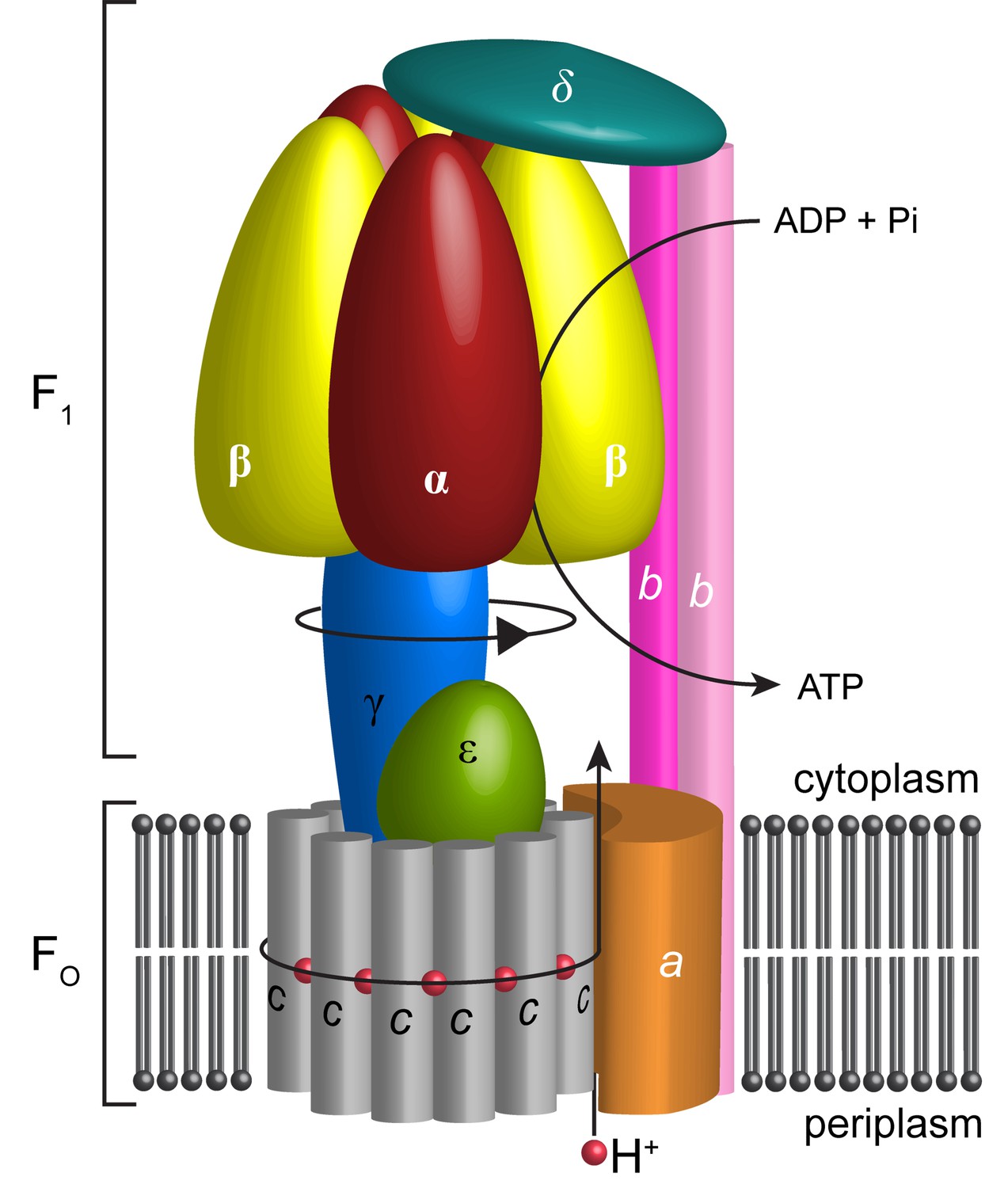
Schematic illustration showing the arrangement of subunits in E. coli F-ATPase.
Subunits α in red, β in yellow, γ in blue, ε in green, c in grey, a in orange, b in magenta or pink, and δ in teal. The proton path and ATP synthesis are labeled accordingly.
The Fo motor is constructed from subunits a, b and c (Figure 1). Subunit c assembles into a ring, thought, in E. coli, to have decameric stoichiometry (Jiang et al., 2001; Ballhausen et al., 2009; Düser et al., 2009; Ishmukhametov et al., 2010), whereas subunits a and b associate to form a helical bundle adjacent to this ring. Recent sub nanometer electron cryo-microscopy (cryo-EM) reconstructions of F-type (Allegretti et al., 2015; Zhou et al., 2015; Kühlbrandt and Davies, 2016; Hahn et al., 2016) and the analogous V- and A-type ATPases (Zhao et al., 2015; Schep et al., 2016) as well as a low-resolution crystal structure of Paracoccus denitrificans F-ATPase (Morales-Rios et al., 2015) are consistent with a two half-channel mechanism for the generation of rotation within the membrane (Vik and Antonio, 1994; Junge et al., 1997). All structures confirm that a four-helix bundle of subunit a, inclined by 20–30˚ to the membrane plane, forms a crucial structural component. In this mechanism, protons from the bacterial periplasm access a conserved negatively charged carboxylate in subunit c (Asp61 in E. coli [Hoppe et al., 1982]) through an aqueous half channel at the subunit a/c interface (Steed and Fillingame, 2008). Neutralizing this carboxylate enables the c-ring to rotate within the hydrophobic membrane and to access a second aqueous half channel that opens to the cytoplasm into which the protons are released (Pogoryelov et al., 2010). A conserved arginine residue in helix-4 of subunit a (Arg210 in E. coli) prevents the c-ring rotating in the opposite direction and short-circuiting of the system (Lightowlers et al., 1987; Cain and Simoni, 1989; Mitome et al., 2010). The sequential binding of protons in combination with thermal fluctuations generates rotation within the complex in a manner akin to a turbine (Oster and Wang, 1999; Pogoryelov et al., 2010; Aksimentiev et al., 2004). The torque generated in the Fo motor is then transferred to the F1 motor by the central shaft consisting of subunits γ and ε (Wilkens et al., 1995). The N- and C-termini of subunit γ form a curved coiled-coil that extends into the central cavity of F1.
The F1 motor is the chemical generator in which ATP is synthesized. The motor comprises a ring of three heterodimers, each containing an active site at the interface of subunits α and β. Within the F1 motor, each αβ dimer has a different conformation at any point in time and can be either empty, bound to ADP and Pi, or bound to ATP (open, half-closed, closed) (Abrahams et al., 1994; Yoshida et al., 2001). These different catalytic states relate to the position of the curved coiled-coil of subunit γ in the central stalk, which drives the conformational changes associated with catalysis. To enable the central stalk to rotate relative to the F1 αβ heterodimers, the Fo and F1 motors need to be coupled. This coupling is mediated by the peripheral stalk that is constructed from subunits b and δ (Figure 1). Subunit b forms an amphipathic homodimeric coiled-coil that spans the periphery of the complex linking subunit a with subunit α, whereas subunit δ provides additional coupling of the C-termini of the b and α subunits (Carbajo et al., 2005; Wilkens et al., 2005).
The bacterial F-ATPase can also function in reverse, employing ATP hydrolysis to generate a proton gradient across the membrane when needed (von Ballmoos et al., 2008). In vivo, subunit ε is believed to change conformation in an ATP-dependent manner to prevent rotation of the complex (Capaldi et al., 1992; Rodgers and Wilce, 2000; Yagi et al., 2007; Imamura et al., 2009) thereby conserving ATP when its concentration is low. This regulatory function is mediated by the C-terminal domain of subunit ε (εCTD) that, when not bound to ATP, opens to an extended conformation and inserts into the αβ heterodimers. The crystal structures of both the E. coli and Bacillus PS3 F1 motors in this autoinhibited state show the εCTD intercalating into the αβ heterodimers (Cingolani and Duncan, 2011; Shirakihara et al., 2015). However, in each structure, the F1 motor had been captured in a different conformation (E. coli – half-closed, closed, open [Cingolani and Duncan, 2011] and Bacillus PS3 – open, closed, open [Shirakihara et al., 2015]) which could either relate to inter species differences or crystal contacts and crystallization conditions.
The crystal structure of the F-ATPase from P. denitrificans (Morales-Rios et al., 2015), that is closely related to E. coli (38% sequence identity over all subunits), shows a similar overall architecture to bovine F1Fo ATP synthase as well as to the main features of A/V ATPases. However, it is inhibited by the ζ-protein rather than by subunit ε, which is generally employed by bacterial F-ATPases for this purpose. Moreover, this crystal structure shows only one conformation of the rotary catalytic cycle.
Here, we present three cryo-EM maps along with molecular models of E. coli F-ATPase in its autoinhibited state, determined to resolutions of 6.9, 7.8, and 8.5 Å. In all three reconstructions, the εCTD is in an extended conformation, stabilizing an overall F1 motor conformation similar to that seen in the thermophilic Bacillus PS3 F1 ATPase structure. Density for the peripheral stalk extends the entire length of the complex and its coiled-coil bifurcates towards the N-terminus to enter the membrane as two separate helices that clamp the a subunit to the c ring. Moreover, our maps allowed us to interpret the complete F1-delta interface, showing the three α subunit N-termini in distinct orientations. Each map also confirmed the c-ring stoichiometry to be decameric, which to date has been only characterized by crosslinking and single molecule analyses. We used our maps in combination with published crosslinking and mutagenesis information to generate a molecular model of the complex in three states. These models provide crucial structural information on a key complex that extends our understanding of the mechanism of rotary ATPases in general, together with information on the bacterial ATP synthase, which is seen as an important antimicrobial target in organisms related to E. coli such as Mycobacterium tuberculosis (Ahmad et al., 2013).
Results
Complete molecular models of three different F-ATPase conformations
Cysteine-free E. coli F-ATPase, as described in Ishmukhametov et al. (2005) where all 10 cysteines were replaced with alanines and a His-tag introduced on the β subunit, was solubilized in digitonin detergent and purified as described in the Materials and methods. This procedure provided pure protein (Figure 2—figure supplement 1) capable of ATP hydrolysis-driven proton pumping upon reconstitution into proteoliposomes (Figure 2—figure supplement 1). N,N`-dicyclohexylcarbodiimide (DCCD), a compound which selectively modifies Asp61 of subunit c at 50 µM (Pogoryelov et al., 2010) completely abolished proton pumping (Figure 2—figure supplement 1B) and inhibited 90% of ATPase activity of isolated protein (Figure 2—figure supplement 1C). Such inhibition indicates coupling between the F1 and Fo motors (Cook et al., 2003; Peskova and Nakamoto, 2000; Tsunoda et al., 2000).
Protein was further examined by cryo-EM without addition of nucleotides. 395,140 particles were picked, of which 216,711 were used in refinement. Three different conformations of the complex were identified using 3D classification in RELION (Scheres, 2012). The particles in each subset were then refined to generate sub-nanometre reconstructions, to a resolution of 6.9, 7.8 and 8.5 Å (Figure 2A–C, and Figure 2—figure supplements 2 and 3). In these three conformations, the central stalk was progressively rotated 120° relative to the peripheral stalk.
Figure 2 with 10 supplements see all
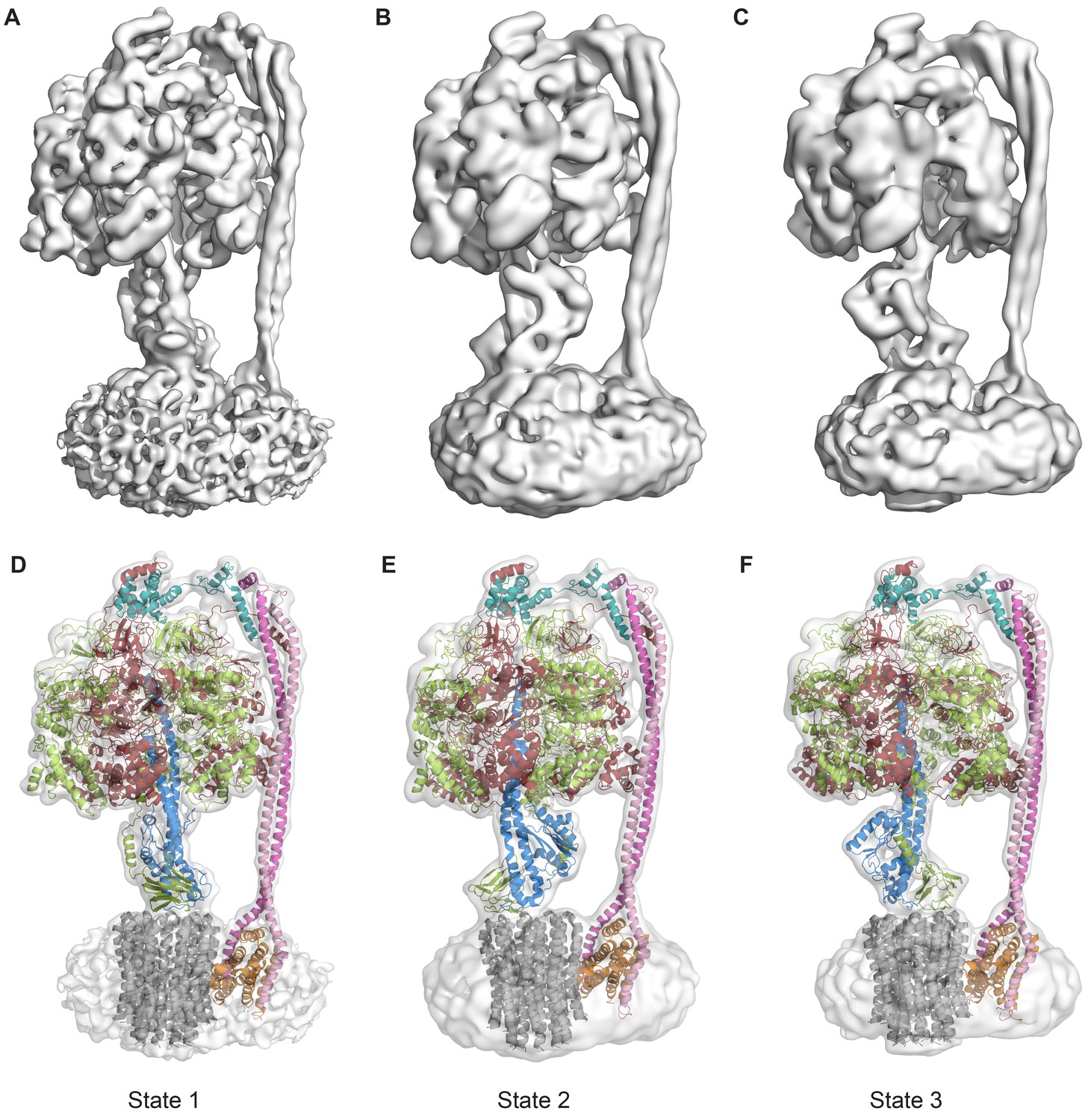
The three states of the autoinhibited E. coli F-ATPase.
(A–B) Cryo-EM maps shown as surface representation, states 1, 2 and 3, respectively, resulting from rotation of the central stalk by 120°. (D–F) Molecular models built into the cryo-EM maps shown as cartoon representation. Subunits α in red, β in yellow, γ in blue, ε in green, c in grey, a in orange, b in magenta or pink and δ in teal.
-
Figure 2—source data 1
Data collection and image processing statistics.
- https://doi.org/10.7554/eLife.21598.005
Even though the resolution of the reconstructions varied throughout the complex, it was sufficient to resolve individual helices. Additional density of the N-terminal His-tag of the β subunit, as well as helical and β sheet patterns observed in parts of the map in the F1 motor region illustrate the high quality of the maps, with the c-ring density being poorest (Figure 2—figure supplement 4). Local resolution estimates showed the region corresponding to the F1 motor to be of highest quality, the Fo motor with moderate detail and, the detergent micelle being clearly the worst region of the map (Figure 2—figure supplement 5). Docking of high-resolution crystal and NMR models of different components into the maps followed by manual building and refinement enabled virtually complete molecular models of the three different states to be built (Figure 2D–F), with varying quality of the docked structures as indicated in Figure 2—figure supplement 6. The positions of the Cys-Ala mutants are depicted in Figure 2—figure supplement 7.
The cryo-EM maps provided novel insights into the architecture and function of the E. coli F-ATPase. Thus, although its overall architecture was similar to that of F-ATPase from P. denitrificans (Morales-Rios et al., 2015) and F1Fo ATP synthases from Bos Taurus (Zhou et al., 2015), Yarrowia lipolytica (Hahn et al., 2016) and Polytomella (Allegretti et al., 2015), with the catalytic F1 motor attached to a proton powered membrane Fo motor and single central and peripheral stalks, differences in the individual motors and peripheral stalk were apparent. Comparison with the membrane-embedded motors from other sub nanometre cryo-EM maps indicated the E. coli F-ATPase had a simpler stator architecture, containing only seven helices in the a and b subunits rather than the eight seen in mitochondrial F-type ATP synthase (Figure 2—figure supplement 8), consistent with labeling approaches (Wada et al., 1999). This difference suggested that the extra helices present in other rotary ATPase subtypes could have additional functions such as the dimerization seen in mitochondria (Hahn et al., 2016).
A movie generated by interpolation between the three states (Video 1) indicated that the F1 motor rocks or wobbles during the catalytic cycle (Kinosita et al., 2000; Stewart et al., 2012) as previously predicted, although of course the structures described here do not represent the complex in its uninhibited active synthesizing form. Two pivot points, one near the peripheral stalk/Fo interface (~bArg36) (Welch et al., 2008) and one near the peripheral stalk/F1 interface (~bGln106), enabled the stalk to accommodate this eccentric movement of F1 (Figure 2—figure supplement 9).
Video 1
Interpolation between States 1, 3 and 2 to simulate ATP synthesis by E. coli F-ATPase.
A and B are rotated 90° about the y-axis.
Subunit δ interaction with subunit b dimer and all three α subunits
The maps showed a long right-handed coiled-coil dimer generated by the two b subunits of the peripheral stalk together with the globular δ subunit that anchors them to the catalytic head (Figure 3A). The quality of the map was sufficient to enable almost the entire of the δ subunit to be built as a polyalanine model (Figure 2D–F), whereas previous structural information was limited to the N-terminal domain (Wilkens et al., 2005). Interestingly, the peripheral stalk contacted all three α subunits via their N-terminal helices, but did so asymmetrically employing three different interfaces with each α subunit (Figure 4). Although the resolution of the map was insufficient to assign the precise interface, the binding of the peripheral stalk to three anchor points in different geometries would provide a molecular key that would result in the δ subunit binding in a single orientation across the top of the symmetrical αβ heterodimers.
Figure 3 with 2 supplements see all

The peripheral and central stalks of E. coli F-ATPase.
(A) The peripheral stalk is comprised of a globular head (subunit δ in teal) and a homodimeric coiled-coil (subunits b in pink and magenta) that bifurcates at the membrane interface to brace subunit a (orange). (B) The εCTD is in an extended conformation, inhibiting the enzyme from rotating. The arrow depicts the extended vs closed conformation of subunit ε.
Figure 4

Subunit δ and peripheral stalk attachments to the α subunits.
Top panel; left, the segmented cryoEM map viewed from the side and right, viewed from above with the orientation of views 1, 2 and 3 depicted. Bottom panel; detailed views of the three attachment points labeled 1, 2 and 3, with δ in teal, b in pink and magenta and α in red.
The peripheral stalk bifurcates into the membrane
The b subunits formed a homodimeric coiled-coil that spaned almost the entire complex (212 of 232 Å) with their N-termini bifurcating just above the membrane to generate two separate helices within the membrane (Figure 3A). This was unexpected, albeit reminiscent of the yeast F-type ATP synthase dimer (Figure 2—figure supplement 8C), where subunit eight is an evolutionary derivative of the bacterial b subunit (Hahn et al., 2016). Furthermore, NMR analysis of the transmembrane domain of E. coli F-ATPase b subunit (Dmitriev et al., 1999) showed a helical structure that was interrupted by a rigid 20° bend at residues 23–26 that result in a structure consistent with the b subunit bifurcation.
Inhibition of the E. coli F-ATPase by central stalk subunit ε
All three reconstructions showed the complex in its autoinhibited state, with clear density for the εCTD extending deep into the central cavity of the F1 enzyme (Figure 3B). Fitting of the E. coli α3β3γε crystal structure (Cingolani and Duncan, 2011) into our cryo-EM maps showed that the β1 subunit had adopted a different more open conformation (Figure 3—figure supplement 1). In the above crystal structure, the εCTD contacts more subunits in F1 (α1, α2, β1, β2 and γ) compared to our cryo-EM reconstructions, where it contacted fewer subunits (α1, α2, β2 and γ). The conformation of our cryo-EM structure was more similar to that seen in the Bacillus PS3 structure (Shirakihara et al., 2015), where it is proposed to be in an ‘open, closed, open’ conformation (Figure 5).
Figure 5

Autoinhibted E. coli F1-ATPase conformation.
The αβ hetrodimers of state 1 as viewed from the membrane with the peripheral stalk to the left of the figure. The ‘open, closed, open’ conformation of the F1 motor is labeled and the positions of nucleotides are shown as blue surfaces.
To ascertain the enzymatic state of the F1 motor, we generated a difference density map between the cryo-EM density and our built model. Remarkably, clear peaks were seen at the nucleotide binding pockets, indicating that the non-catalytic binding sites in subunit α contained nucleotide, as well as the ‘closed’ αβ heterodimer (Figure 5). These were modeled as ATP and ADP respectively based on known orientations of these nucleotides from high-resolution crystal structures. Interestingly, this did not correspond to the nucleotide binding states of either the E. coli (Cingolani and Duncan, 2011) or the PS3 crystal structures (Shirakihara et al., 2015).
Single particle analysis revealed highly uniform homogeneity of the protein preparation, where 100% of F1Fo molecules observed demonstrated the extended conformation of εCTD. Our structural data were supported by an enzymatic assay (Figure 3—figure supplement 2), where the extent of ATP hydrolysis inhibition of the protein by subunit ε was tested with N,N-dimethyldodecylamine N-oxide (LDAO), a well-known activator of the subunit ε inhibited protein. LDAO used at 0.4% (weight/volume) concentration has been shown to stimulate ATP hydrolysis by the E coli protein 3–4 times (Dunn et al., 1990; Peskova and Nakamoto, 2000), including earlier studies (Ishmukhametov et al., 2008, 2016) on the same cysteine-free F1Fo construct using the same batch of LDAO. Prior to addition of LDAO, the hydrolysis rate was 0.75 µmol ATP/min/mg protein but surprisingly in presence of 0.4% LDAO it was stimulated ~13 times. To our best knowledge, such a high LDAO stimulation of ATP hydrolysis by E. coli F1Fo was not described in the literature before and is consistent with our single particle data.
The E. coli Fo motor – architecture of a proton channel
Density in Fo defined the overall architecture of the membrane-embedded motor together with two invaginations of the detergent micelle that have previously been proposed to facilitate proton translocation (Allegretti et al., 2015; Kühlbrandt and Davies, 2016) (Figure 6—figure supplement 1). While the overall density of the c-ring was relatively weak, 10 peaks of density were clearly present when viewed from above (Figure 6—figure supplement 2), confirming the stoichiometry of the c-ring to be decameric in E. coli F-ATPase (Jiang et al., 2001; Ballhausen et al., 2009; Düser et al., 2009; Ishmukhametov et al., 2010). Furthermore, density inside the c-ring corroborates data suggesting it to be filled with phospholipids (Oberfeld et al., 2006).
By combining the helical density from the cryo-EM maps (Figure 6A), with models previously suggested for the related bovine subunit, together with crosslinking data and transmembrane topography prediction for the E. coli F-ATPase (Jiang and Fillingame, 1998; Valiyaveetil and Fillingame, 1998; Moore and Fillingame, 2008; Wada et al., 1999), it was possible to build a molecular model of the a subunit (Figure 6B). The crosslinks mapped to two clusters (Figure 6—figure supplement 3), allowing a likely sequence register for the model to be proposed. This was consistent with the two half channel hypothesis, placing Arg210 of subunit a adjacent to Asp61 of the c-ring (Figure 6B). Interestingly, density for the c subunit is clearest adjacent to Arg210 of subunit a suggesting this area to be well ordered (Figure 6—figure supplement 4).
Figure 6 with 5 supplements see all
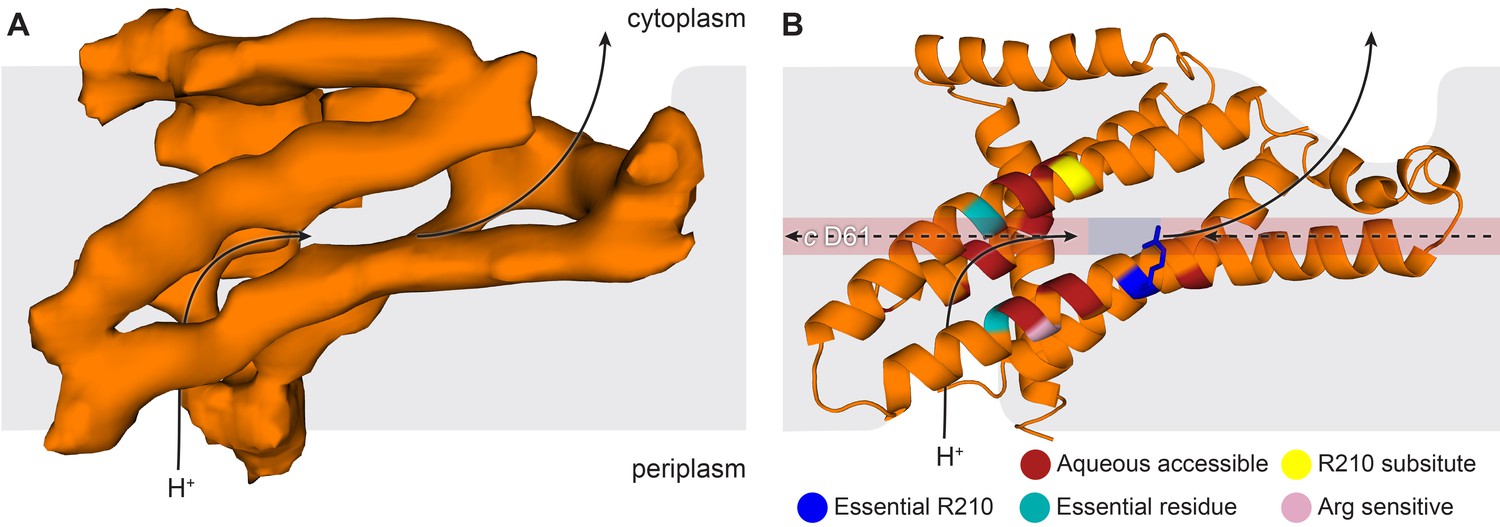
The E. coli F-ATPase subunit a and the suggested path of proton translocation.
(A) Density map of subunit a, shown as orange surface viewed from the c-ring. Grey outline depicts invaginations of the detergent micelle, with arrows showing possible proton path. (B) Cartoon representation of subunit a with a horizontal stripe to depict the position of Asp61 on the c-ring (red where Asp61 would be bound to a proton and blue when bound to Arg210). Functional mutants labeled as follows; essential arginine in blue, substitution with Arg210 resulting in functional complex in yellow, mutation to arginine resulting in a dysfunctional complex in teal and residues that are aqueous accessible in red. Solid arrows show a possible proton path via two ‘half’ channels and dashed arrows show the path when bound to Asp61 of the c-ring and rotating.
Discussion
We have generated cryo-EM maps of a bacterial F-ATPase providing new insights into this rotary ATPase subtype. These maps enabled the generation of a molecular model that presents a framework onto which the vast array of information available on the widely studied E. coli enzyme can be mapped, including the attachment of the peripheral stalk to the F1 and Fo motors, the inhibition mediated by the ε subunit, and the stoichiometry of the c-ring. In addition, the model confirmed the presence of key features such as the near ‘horizontal’ helices angled at 20–30° relative to the membrane, indicating that this feature is conserved and is a signature of F, V and A-type ATPases.
Our reconstructions extended previous work (Wilkens and Capaldi, 1998b) by showing the structure of the complete peripheral stalk and how it is attached to both the F1 and Fo components. The peripheral stalk functions to counteract rotation of the F1 stator relative to the Fo stator as the central stalk rotates, but must also accommodate conformational changes in the F1 motor during catalysis. The peripheral stalk is based on a long right-handed coiled-coil dimer, that is the hallmark of all rotary ATPase peripheral stalks (Lee et al., 2010), showed near parallel α-helices, based on an 11-residue hendecad sequence repeat, spanning the space between the Fo and F1 motors, that changed into a 15-residue quindecad sequence repeat along the F1 motor enabling it to accommodate conformational changes (Stewart and Stock, 2012; Stewart et al., 2012). Although sequence identity is low (22%), the overall fold of the soluble portion of the peripheral stalk was strikingly similar to that of Thermus thermophilus A-ATPase (Lee et al., 2010), illustrating a strong evolutionary pressure for this fold and its function to prevent rotation between the αβ heterodimers and the a subunit while accommodating wobbling of the F1 motor. The bifurcation of the peripheral stalk coiled-coil into two separate helices in the membrane was unexpected, but this arrangement enabled the peripheral stalk to bind to the a subunit in two regions. This in turn increased the distance about the fulcrum of interaction, which may help clamp the a subunit to the c-ring and counteract rotation and pivoting relative to the rotor. The cryo-EM maps also indicated that the peripheral stalk is able to flex about two hinges adjacent to the F1 and Fo motors, enabling it to accommodate conformational changes in the catalytic head.
In addition to its fundamental importance in cell metabolism, the regulation of ATP synthase is also an attractive antibiotic discovery target for pathogenic bacteria closely related to E. coli (Ahmad et al., 2013). Bacterial F-ATPases employ a unique method of regulation whereby the enzyme can be autoinhibited with the integral subunit ε. In all three rotational states of the E. coli F-ATPase, the εCTD had an extended conformation, albeit with a different proportion of particles observed at each state (46%, 30% and 24%) (Figure 2—figure supplement 3), suggesting State one to be the lowest energy. No reconstruction at any stage of data processing contained density corresponding to a closed/down conformation of the εCTD, and this along with the strong stimulation of ATPase activity by LDAO, suggests that the majority of the protein to be in an autoinhibited form. Although the position of εCTD relative to the αβ subunits was similar to that of the mitochondrial inhibitor protein (IF1), the observation that it bound to all three states was different to that seen for IF1 that is bound to a single rotational F1 state (α/βDP site proximal to the peripheral stalk) in the F1Fo ATP synthase dimer structure (Hahn et al., 2016). The cryo-EM maps resembled the ‘open, closed, open’ conformation as seen in the Bacillus PS3 F1 crystal structure, despite different nucleotide binding positions. However, the major contacts formed by the F1 motor with the εCTD in our maps were similar to that of the E. coli crystal structure, except that one β subunit changed conformation substantially (Figure 3—figure supplement 1). Because our cryo-EM study was performed in the absence of externally added nucleotide, it is likely that the structures correspond to the autoinhibited conformations in solution, and the crystal structure of the isolated E. coli F1 could instead represent a partially bound state, especially since the crystals were soaked in 1 mM AMPPNP prior to freezing (Cingolani and Duncan, 2011).
The c-ring is responsible for the rotation of the complex and contains the conserved carboxylate that binds the proton. Different species have varying numbers of subunits in their ring, believed to ‘gear’ the motor tailoring them to their environment and ranging from 8 to 15 subunits (Stock et al., 1999; Pogoryelov et al., 2007; Watt et al., 2010; Stewart et al., 2013). Although the density corresponding to the c-ring was quite weak, 10 peaks can be discriminated in the density at either end of the ring (Figure 6—figure supplement 2). This confirmed the c-ring stoichiometry of E. coli F-ATPase that had previously been suggested to be decameric by crosslinking (Ballhausen et al., 2009), fusion (Jiang et al., 2001) and single molecule analysis (Düser et al., 2009; Ishmukhametov et al., 2010).
The model of subunit a generated from our cryo-EM maps confirmed that the Fo motor likely operates using two half channels separated by a conserved arginine that directs its rotation (Vik and Antonio, 1994; Junge et al., 1997). Importantly, in this context, our model placed Arg210, which is believed to mediate the rotation of the c-ring, adjacent to the conserved carboxylate residue, Asp61, of the c subunit that has been shown to bind protons (Vik and Antonio, 1994; Pogoryelov et al., 2010) (Figure 6b and Video 2). In addition Gln252, which can be substituted with Arg210 and retain ATP synthase function (Ishmukhametov et al., 2008; Hatch et al., 1995), is positioned on a proximal helix with a similar distance to Asp61 of subunit c (Figure 6B and Figure 6—figure supplement 5). Moreover, mainly charged residues, that have been shown to be aqueous accessible (Angevine and Fillingame, 2003; Angevine et al., 2003), map to two channel-like areas exposed to solvent by the invagination of the detergent micelle (Figure 6B and Figure 6—figure supplement 5). Furthermore, residue A217, which has been shown to be sensitive to arginine mutation, and therefore been suggested to be near or part of the aqueous pocket (Cain and Simoni, 1989), is positioned next to the periplasmic half channel (Figure 6B and Figure 6—figure supplement 5). Additionally. residues E219 and H245, which when substituted for one another result in a functional enzyme, are proximal to one another in our model (Cain and Simoni, 1988) (Figure 6—figure supplement 5). Further analysis of inter subunit crosslinking fits our model well (Figure 6—figure supplement 3), with the distances between the c-ring and a subunit being minimal.
Video 2
View of Fo motor during ATP synthesis.
Same as main text Figure 6b, but with rotating c-ring in the foreground.
In summary, our models show a new level of detail for the bacterial F-ATPase, providing a template for further experiments as well as to guide future antibiotic discovery in related pathogenic bacteria.
Accession codes
The three models and maps were deposited in the pdb and emDB with codes 5T4O, EMD-8357 (State 1), 5 T4P, EMD-8358 (State 2), 5T4Q and EMD-8359 (State 3).
Materials and methods
Protein purification
Request a detailed protocolA cysteine-free version of E. coli F-ATPase cloned in plasmid pFV2 and expressed in E. coli DK8 strain was used (Ishmukhametov et al., 2005). Cells were grown at 37°C in LB medium supplemented with 100 μg/ml ampicillin, for 4–5 hr. The harvested cells were resuspended in lysis buffer containing 50 mM Tris/Cl pH 8.0, 100 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 2.5% glycerol and 1 μg/ml DNase I and processed by one pass in French press at 20 kPSI. Cellular debris was removed by centrifuging at 7700 × g for 15 min, and the membranes were collected by ultracentrifugation at 100,000 × g for 1 hr. The ATP synthase complex was extracted from membranes at 4°C for 1 hr by resuspending the pellet in extraction buffer consisting of 20 mM Tris/Cl, pH 8.0, 300 mM NaCl, 2 mM MgCl2, 100 mM sucrose, 20 mM imidazole, 10% glycerol, 4 mM digitonin and EDTA-free protease inhibitor tablets (Roche). The complex was then purified by binding on Talon resin (Clontech) and eluted in 150 mM imidazole. The protein was further purified and sugars removed by size exclusion chromatography on a 16/60 Superose six column equilibrated in a buffer containing 20 mM Tris/Cl pH 8.0, 100 mM NaCl, 4 mM digitonin and 2 mM MgCl2. The purified protein was then concentrated to 2 mg/ml for cryo-EM.
Protein reconstitution into proteoliposomes
Request a detailed protocolSeventy microgram of F1Fo was reconstituted into extrusion-preformed 100 nm soybean phosphatidylcholine liposomes exactly as descried (Ishmukhametov et al., 2016).
Functional assays
Request a detailed protocolProton pumping by proteoliposomes was studied using quenching of a pH sensitive fluorescent probe 9-Amino-6-Chloro-2-Methoxyacridine (ACMA) exactly as described (Ishmukhametov et al., 2016). The assay was performed with 100 µl of proteoliposomes in 2-ml cuvettes. The reaction was started with 0.25 mM ATP and stopped by 2 µM of the uncoupler FCCP.
ATP hydrolase activity and its stimulation by LDAO was measured with ATP regenerating system using 5 µg of the protein with 1 mM ATP exactly as described (Ishmukhametov et al., 2016).
DCCD inhibition of proteoliposomes was done as described (Ishmukhametov et al., 2016). DCCD inhibition of pure F1Fo was done as described (Ishmukhametov et al., 2005), with the following modification. Ten microgram of the protein was incubated in 1 ml buffer A (50 mM MES, pH 6.4, 100 mM KCl, 1 mM MgCl2) with 50 µM DCCD for 30 min at room temperature. Control sample contained 1% ethanol instead of DCCD. Reaction was started by mixing the inhibited protein with 1 ml of buffer A containing all the components of ATP regenerating system.
All the functional experiments presented here were repeated two to three times using the protein isolated by MS and shipped to RI at liquid N2 temperature. Results of typical experiments are shown.
Cryo-EM grid preparation and data collection
Request a detailed protocolAliquots of 4 μl of purified E. coli F-ATPase at a concentration of 3.58 μM were placed on glow-discharged holey carbon grids (Quantifoils Copper R2/2, 200 Mesh). Grids were blotted for 2 s and flash-frozen in liquid ethane using an FEI Vitrobot Mark IV. Grids were transferred to an FEI Titan Krios transmission electron microscope that was operating at 300 kV. Images were recorded automatically using the FEI EPU software, yielding a pixel size of 1.4 Å. A dose rate of 29 electrons (spread over 20 frames) per Å2 per second, and an exposure time of 2 s were used on the Falcon-II detector. 8640 movies were collected.
Data processing
Request a detailed protocolMotionCorr (Li et al., 2013) was used to correct local beam-induced motion and to align resulting frames. Defocus and astigmatism values were estimated using CTFFIND4 (Rohou and Grigorieff, 2015), and 252 micrographs were excluded due to drift or excessive ice contamination. 1208 particles were manually picked and subjected to 2D classification to generate templates for autopicking in RELION (Scheres, 2012). The automatically picked micrographs were manually inspected to remove false positives, finally yielding 395,140 particles. These particles then underwent two rounds of 2D classification to generate 22 classes with 311,887 particles. The final particles were classified into four 3D classes using a previously generated model from a low-resolution data set of the same sample (unpublished), low-pass filtered to 60 Å. The resolution was estimated using Fourier Shell Correlation (FSC = 0.143, gold-standard). Three of the four classes containing 104,510 (State 1), 67,829 (State 2) and 53,587 (State 3) particles were movie-refined and post-processed in RELION producing maps at 7.4, 7.8, 8.5 Å, respectively (Figure 2—figure supplement 10). State 1 was further processed using masked classification (Bai et al., 2015) with residual signal subtraction with a mask created by removing parts of the detergent micelle. Three out of the four classes from this classification containing 95,345 particles were combined and refined to generate the final 6.9 Å map. Figure 2—figure supplement 3 is a summary of these methods, shown as a flowchart. Local resolution of different parts of the complex was estimated using RELION and ResMap (Kucukelbir et al., 2014).
Model building
Request a detailed protocolCrystal and NMR structures of subunits from E. coli (αβγε - 3oaa [Cingolani and Duncan, 2011], δ - 2a7u [Wilkens et al., 2005], b - 1b9u [Dmitriev et al., 1999], 1l2p [Del Rizzo et al., 2002] and 2khk [Priya et al., 2009]) and related organisms (c - 3u2f [Symersky et al., 2012] and a - 5fik [Zhou et al., 2015]) were rigid body docked into the highest resolution cryo-EM map and the side chains ‘pruned’ to Cα. The sequence was mapped to subunit a using crosslinks as restraints. Subsequent manual model building and refinement was performed with Coot (Emsley et al., 2010), Phenix (Adams et al., 2010) and Refmac (Murshudov et al., 2011) (excluding subunit c due to weak density), with crosslinks again used as external restraints (a summary of the types of models used to build the initial model can be found in Figure 2—figure supplement 6). Nucleotide occupancy was determined by first building the model without any nucleotide present, and then segmenting the map and selecting any density with 15% overlap with atoms and deleting this density. Nucleotide was subsequently docked into this difference density using the known positions from previous structures. Once a complete model was built of the highest resolution map, this was docked and refined to the other two maps to create three models. The three models and maps were deposited in the pdb and emDataBank with codes 5T4O, EMD-8357 (State 1), 5 T4P, EMD-8358 (State 2), 5T4Q and EMD-8359 (State 3). Data statistics shown in Figure 2—source data 1.
Data availability
-
Autoinhibited E. coli ATP synthase state 1Publicly available at the RCSB Protein Data Bank (accession no. 5T4O).
-
Autoinhibited E. coli ATP synthase state 1Publicly available at the Protein Data Bank in Europe (accession no. EMD-8357).
-
Autoinhibited E. coli ATP synthase state 2Publicly available at the RCSB Protein Data Bank (accession no. 5T4P).
-
Autoinhibited E. coli ATP synthase state 2Publicly available at the Protein Data Bank in Europe (accession no. EMD-8358).
-
Autoinhibited E. coli ATP synthase state 3Publicly available at the RCSB Protein Data Bank (accession no. 5T4Q).
-
Autoinhibited E. coli ATP synthase state 3Publicly available at the Protein Data Bank in Europe (accession no. EMD-8359).
References
-
PHENIX: a comprehensive Python-based system for macromolecular structure solutionActa Crystallographica Section D Biological Crystallography 66:213–221.https://doi.org/10.1107/S0907444909052925
-
ATP synthase: a molecular therapeutic drug target for antimicrobial and antitumor peptidesCurrent Medicinal Chemistry 20:1956–1973.https://doi.org/10.2174/0929867311320150003
-
Insights into the molecular mechanism of rotation in the Fo sector of ATP synthaseBiophysical Journal 86:1332–1344.https://doi.org/10.1016/S0006-3495(04)74205-8
-
Aqueous access channels in subunit a of rotary ATP synthaseJournal of Biological Chemistry 278:6066–6074.https://doi.org/10.1074/jbc.M210199200
-
Constant c10 ring stoichiometry in the Escherichia coli ATP synthase analyzed by cross-linkingJournal of Bacteriology 191:2400–2404.https://doi.org/10.1128/JB.01390-08
-
Impaired proton conductivity resulting from mutations in the a subunit of F1F0 ATPase in Escherichia coliThe Journal of Biological Chemistry 261:10043–10050.
-
Interaction between Glu-219 and His-245 within the a subunit of F1F0-ATPase in escherichia coliThe Journal of Biological Chemistry 263:6606–6612.
-
Proton translocation by the F1F0ATPase of escherichia coli. mutagenic analysis of the a subunitThe Journal of Biological Chemistry 264:3292–3300.
-
Structure of the Escherichia coli ATP synthase and role of the gamma and epsilon subunits in coupling catalytic site and proton channeling functionsJournal of Bioenergetics and Biomembranes 24:435–439.https://doi.org/10.1007/BF00762359
-
Structure of the F1-binding domain of the stator of bovine F1Fo-ATPase and how it binds an alpha-subunitJournal of Molecular Biology 351:824–838.https://doi.org/10.1016/j.jmb.2005.06.012
-
Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformationNature Structural & Molecular Biology 18:701–707.https://doi.org/10.1038/nsmb.2058
-
Structure of the membrane domain of subunit b of the Escherichia coli F0F1 ATP synthaseJournal of Biological Chemistry 274:15598–15604.https://doi.org/10.1074/jbc.274.22.15598
-
36 degrees step size of proton-driven c-ring rotation in FoF1-ATP synthaseThe EMBO Journal 28:2689–2696.https://doi.org/10.1038/emboj.2009.213
-
Features and development of CootActa Crystallographica Section D Biological Crystallography 66:486–501.https://doi.org/10.1107/S0907444910007493
-
Ultrafast purification and reconstitution of His-tagged cysteine-less Escherichia coli F1Fo ATP synthaseBiochimica Et Biophysica Acta (BBA) - Bioenergetics 1706:110–116.https://doi.org/10.1016/j.bbabio.2004.09.012
-
ATP synthesis without R210 of subunit a in the Escherichia coli ATP synthaseBiochimica Et Biophysica Acta (BBA) - Bioenergetics 1777:32–38.https://doi.org/10.1016/j.bbabio.2007.11.004
-
ATP synthase: an electrochemical transducer with rotatory mechanicsTrends in Biochemical Sciences 22:420–423.https://doi.org/10.1016/S0968-0004(97)01129-8
-
A rotary molecular motor that can work at near 100% efficiencyPhilosophical Transactions of the Royal Society of London. Series B, Biological Sciences 355:473–489.https://doi.org/10.1098/rstb.2000.0589
-
Quantifying the local resolution of cryo-EM density mapsNature Methods 11:63–65.https://doi.org/10.1038/nmeth.2727
-
Rotary ATPases: A New Twist to an Ancient MachineTrends in Biochemical Sciences 41:106–116.https://doi.org/10.1016/j.tibs.2015.10.006
-
The structure of the peripheral stalk of Thermus thermophilus H+-ATPase/synthaseNature Structural & Molecular Biology 17:373–378.https://doi.org/10.1038/nsmb.1761
-
The proton pore in theEscherichia coli F0F1-ATPase: substitution of glutamate by glutamine at position of the alpha-subunit prevents F0-mediated proton permeabilityBiochimica Et Biophysica Acta 933:241–248.
-
The proton pore in the Escherichia coli F0F1-ATPase: a requirement for arginine at position 210 of the a-subunitBiochimica Et Biophysica Acta (BBA) - Bioenergetics 894:399–406.https://doi.org/10.1016/0005-2728(87)90118-6
-
Structural interactions between transmembrane helices 4 and 5 of subunit a and the subunit c ring of Escherichia coli ATP synthaseJournal of Biological Chemistry 283:31726–31735.https://doi.org/10.1074/jbc.M803848200
-
Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 Å resolutionProceedings of the National Academy of Sciences 112:13231–13236.https://doi.org/10.1073/pnas.1517542112
-
REFMAC5 for the refinement of macromolecular crystal structuresActa Crystallographica Section D Biological Crystallography 67:355–367.https://doi.org/10.1107/S0907444911001314
-
Energy-transducing H+-ATPase of Escherichia coli. Reconstitution of proton translocation activity of the intrinsic membrane sectorThe Journal of Biological Chemistry 255:5643–5648.
-
Microscopic rotary mechanism of ion translocation in the F(o) complex of ATP synthasesNature Chemical Biology 6:891–899.https://doi.org/10.1038/nchembio.457
-
The oligomeric state of c rings from cyanobacterial F-ATP synthases varies from 13 to 15Journal of Bacteriology 189:5895–5902.https://doi.org/10.1128/JB.00581-07
-
Structure of the gamma-epsilon complex of ATP synthaseNature Structural Biology 7:1051–1054.https://doi.org/10.1038/80975
-
CTFFIND4: Fast and accurate defocus estimation from electron micrographsJournal of Structural Biology 192:216–221.https://doi.org/10.1016/j.jsb.2015.08.008
-
RELION: implementation of a Bayesian approach to cryo-EM structure determinationJournal of Structural Biology 180:519–530.https://doi.org/10.1016/j.jsb.2012.09.006
-
Subunit a facilitates aqueous access to a membrane-embedded region of subunit c in Escherichia coli F1F0 ATP synthaseThe Journal of Biological Chemistry 283:12365–12372.https://doi.org/10.1074/jbc.M800901200
-
Rotary ATPases--dynamic molecular machinesCurrent Opinion in Structural Biology 25:40–48.https://doi.org/10.1016/j.sbi.2013.11.013
-
The dynamic stator stalk of rotary ATPasesNature Communications 3:687.https://doi.org/10.1038/ncomms1693
-
Priming a molecular motor for disassemblyStructure 20:1799–1800.https://doi.org/10.1016/j.str.2012.10.003
-
Structure of the c10 ring of the yeast mitochondrial ATP synthase in the open conformationNature Structural & Molecular Biology 19:485–491.https://doi.org/10.1038/nsmb.2284
-
Transmembrane topography of subunit a in the Escherichia coli F1F0 ATP synthaseJournal of Biological Chemistry 273:16241–16247.https://doi.org/10.1074/jbc.273.26.16241
-
A mechanism of proton translocation by F1F0 ATP synthases suggested by double mutants of the a subunitThe Journal of Biological Chemistry 269:30364–30369.
-
Unique rotary ATP synthase and its biological diversityAnnual Review of Biophysics 37:43–64.https://doi.org/10.1146/annurev.biophys.37.032807.130018
-
A novel labeling approach supports the five-transmembrane model of subunit a of the Escherichia coli ATP synthaseJournal of Biological Chemistry 274:17353–17357.https://doi.org/10.1074/jbc.274.24.17353
-
The b (arg36) contributes to efficient coupling in F(1)F (O) ATP synthase in Escherichia coliJournal of Bioenergetics and Biomembranes 40:1–8.https://doi.org/10.1007/s10863-008-9124-3
-
Electron microscopic evidence of two stalks linking the F1 and F0 parts of the Escherichia coli ATP synthaseBiochimica Et Biophysica Acta (BBA) - Bioenergetics 1365:93–97.https://doi.org/10.1016/S0005-2728(98)00048-6
-
ATP synthase--a marvellous rotary engine of the cellNature Reviews. Molecular Cell Biology 2:669–677.https://doi.org/10.1038/35089509
Article and author information
Author details
Alastair G Stewart

Funding
Biotechnology and Biological Sciences Research Council (BB/L01985X/1)
- Robert Ishmukhametov
National Health and Medical Research Council (1004620)
- Daniela Stock
National Health and Medical Research Council (1109961)
- Daniela Stock
National Health and Medical Research Council (1022143)
- Daniela Stock
National Health and Medical Research Council (1047004)
- Daniela Stock
Ministry of Education - Singapore (MOE2012-T3-1-001)
- Sara Sandin
National Health and Medical Research Council (1090408)
- Alastair G Stewart
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
AGS was supported by a National Health and Medical Research Council Fellowship 1090408. DS was supported by National Health and Medical Research Council Fellowships APP1004620 and APP1109961. This work was funded by the National Health and Medical Research Council project grants APP1022143 and APP1047004. ASWW and SS are supported by the Singapore Ministry of Education Academic Research Fund Tier 3 (MOE2012-T3-1-001). R I was supported by BBSRC grant BB/L01985X/1. We thank and acknowledge Daniela Rhodes, the NTU Institute of Structural Biology, the Netherlands Centre for Electron Nanoscopy (NeCEN) at Leiden University, FEI, Sarah Neumann and Max Maletta for help with single particle cryo-EM data collection. NeCEN is supported by the Netherlands Organization for Scientific Research (NWO) and the European Regional Development Fund of the European Commission. The Monash Centre for Electron Microscopy is acknowledged for initial screening of samples.
Copyright
© 2016, Sobti et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 7,344
- views
-
- 1,008
- downloads
-
- 130
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 130
- citations for umbrella DOI https://doi.org/10.7554/eLife.21598
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states
eLife 5:e21598.
https://doi.org/10.7554/eLife.21598




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}